Department of Medical Genome Sciences, Graduate School of Frontier Sciences, University of Tokyo, Tokyo, Japan.
Mol Biol Evol. 2013 Jun;30(6):1454-64. doi: 10.1093/molbev/mst055. Epub 2013 Mar 16.
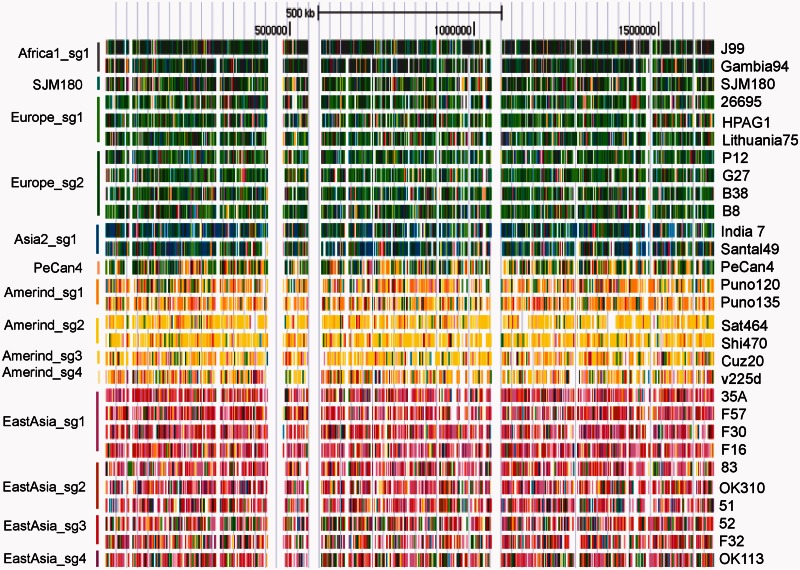
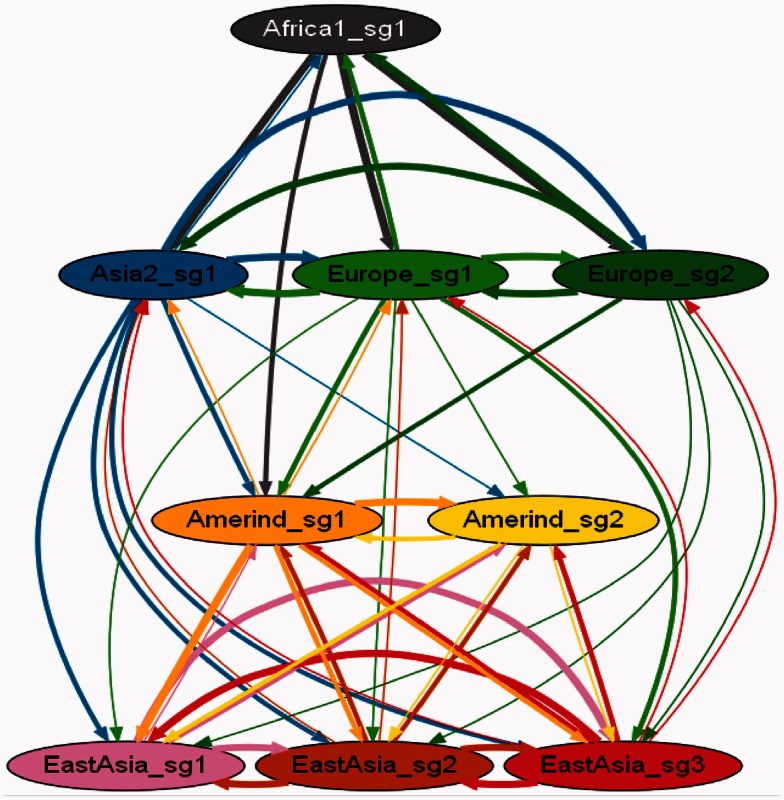
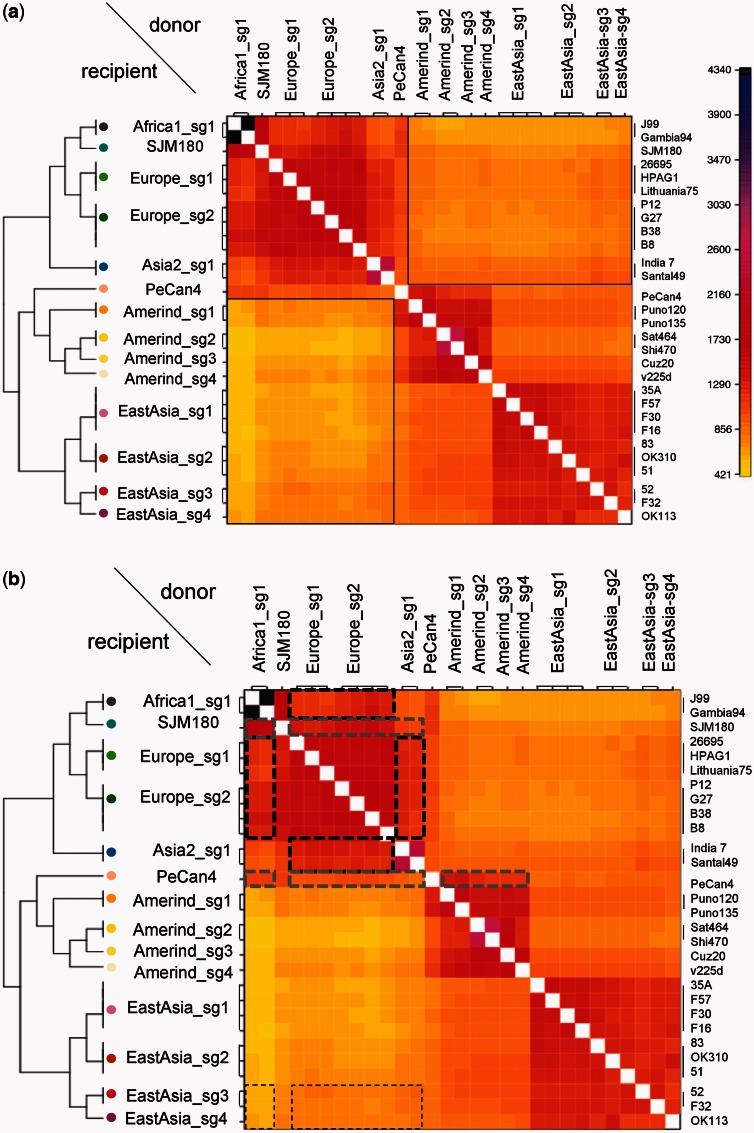
Identifying population structure forms an important basis for genetic and evolutionary studies. Most current methods to identify population structure have limitations in analyzing haplotypes and recombination across the genome. Recently, a method of chromosome painting in silico has been developed to overcome these shortcomings and has been applied to multiple human genome sequences. This method detects the genome-wide transfer of DNA sequence chunks through homologous recombination. Here, we apply it to the frequently recombining bacterial species Helicobacter pylori that has infected Homo sapiens since their birth in Africa and shows wide phylogeographic divergence. Multiple complete genome sequences were analyzed including sequences from Okinawa, Japan, that we recently sequenced. The newer method revealed a finer population structure than revealed by a previous method that examines only MLST housekeeping genes or a phylogenetic network analysis of the core genome. Novel subgroups were found in Europe, Amerind, and East Asia groups. Examination of genetic flux showed some singleton strains to be hybrids of subgroups and revealed evident signs of population admixture in Africa, Europe, and parts of Asia. We expect this approach to further our understanding of intraspecific bacterial evolution by revealing population structure at a finer scale.
确定种群结构是遗传和进化研究的重要基础。目前大多数用于识别种群结构的方法在分析基因组范围内的单倍型和重组方面存在局限性。最近,已经开发出一种基于染色体绘画的计算方法来克服这些缺点,并已应用于多个人类基因组序列。该方法通过同源重组检测全基因组范围内的 DNA 序列片段转移。在这里,我们将其应用于经常重组的细菌物种幽门螺杆菌,该细菌自非洲人类诞生以来就感染了人类,并表现出广泛的系统地理分化。我们分析了多个完整的基因组序列,包括我们最近测序的来自日本冲绳的序列。与仅检查 MLST 管家基因或核心基因组系统发育网络分析的先前方法相比,新方法揭示了更精细的种群结构。在欧洲、美洲印第安人和东亚群体中发现了新的亚群。对遗传通量的研究表明,一些单倍型菌株是亚群的杂种,并揭示了非洲、欧洲和亚洲部分地区人口混合的明显迹象。我们预计这种方法将通过更精细地揭示种群结构来进一步了解种内细菌进化。