Podell Sheila, Gaasterland Terry, Allen Eric E
Marine Biology Research Division, Scripps Institution of Oceanography University of California at San Diego, La Jolla, CA 92093 USA.
BMC Bioinformatics. 2008 Oct 7;9:419. doi: 10.1186/1471-2105-9-419.
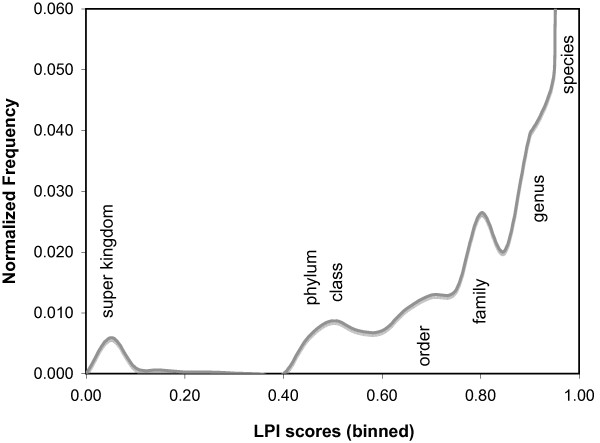
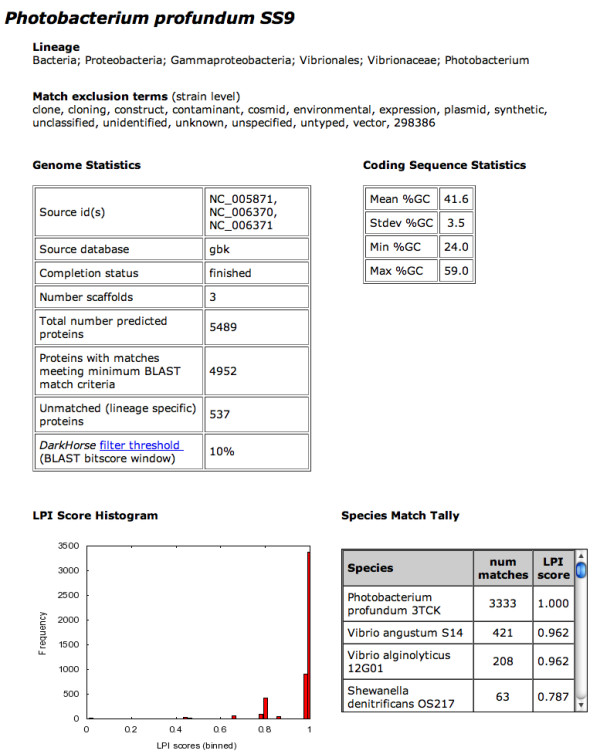
The process of horizontal gene transfer (HGT) is believed to be widespread in Bacteria and Archaea, but little comparative data is available addressing its occurrence in complete microbial genomes. Collection of high-quality, automated HGT prediction data based on phylogenetic evidence has previously been impractical for large numbers of genomes at once, due to prohibitive computational demands. DarkHorse, a recently described statistical method for discovering phylogenetically atypical genes on a genome-wide basis, provides a means to solve this problem through lineage probability index (LPI) ranking scores. LPI scores inversely reflect phylogenetic distance between a test amino acid sequence and its closest available database matches. Proteins with low LPI scores are good horizontal gene transfer candidates; those with high scores are not.
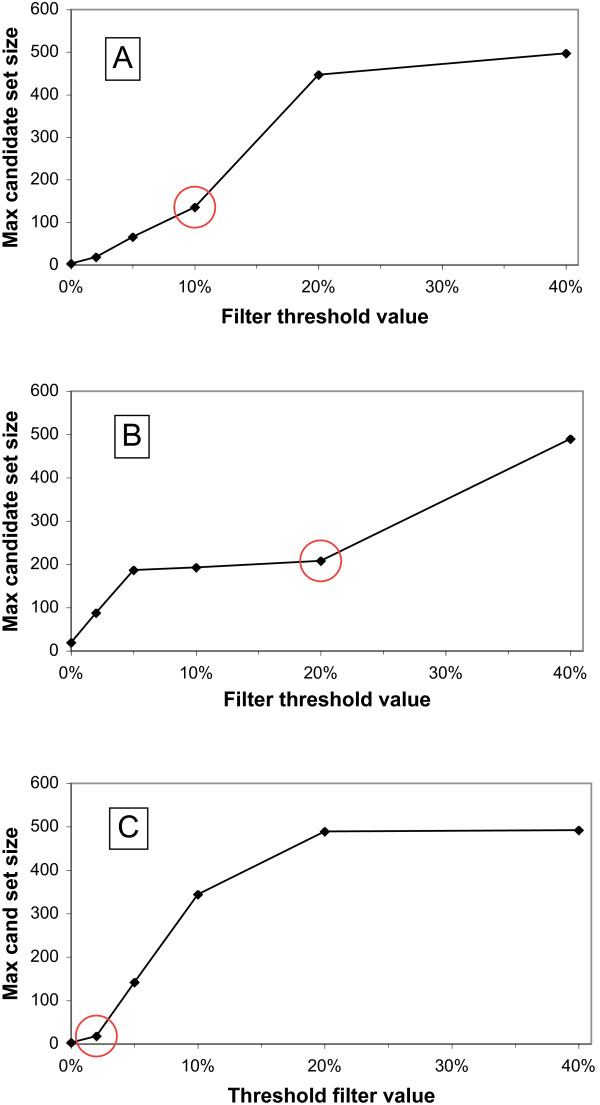
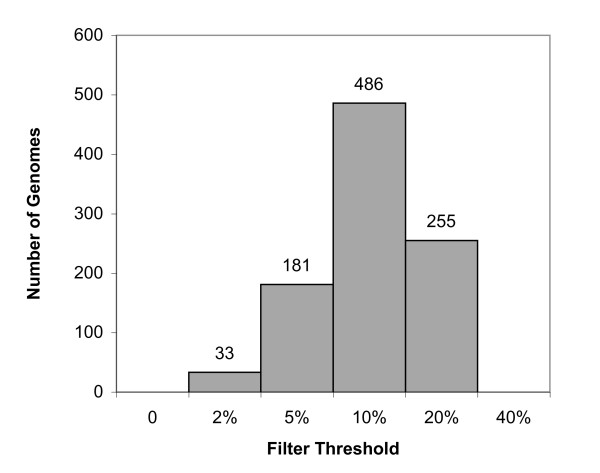
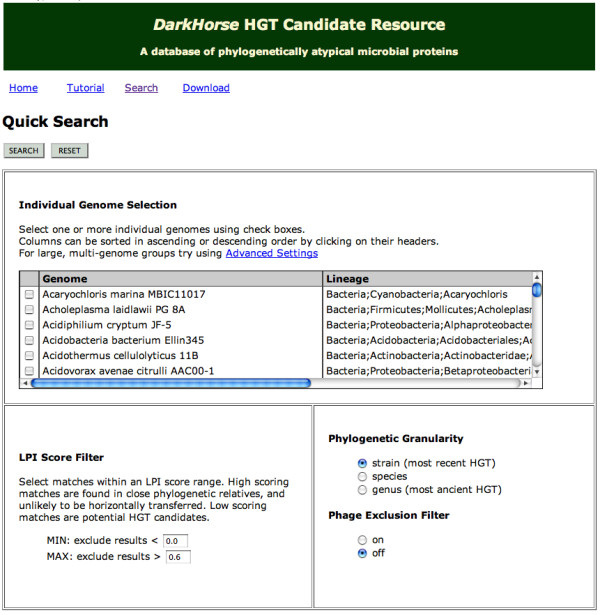
The DarkHorse algorithm has been applied to 955 microbial genome sequences, and the results organized into a web-searchable relational database, called the DarkHorse HGT Candidate Resource http://darkhorse.ucsd.edu. Users can select individual genomes or groups of genomes to screen by LPI score, search for protein functions by descriptive annotation or amino acid sequence similarity, or select proteins with unusual G+C composition in their underlying coding sequences. The search engine reports LPI scores for match partners as well as query sequences, providing the opportunity to explore whether potential HGT donor sequences are phylogenetically typical or atypical within their own genomes. This information can be used to predict whether or not sufficient information is available to build a well-supported phylogenetic tree using the potential donor sequence.
The DarkHorse HGT Candidate database provides a powerful, flexible set of tools for identifying phylogenetically atypical proteins, allowing researchers to explore both individual HGT events in single genomes, and large-scale HGT patterns among protein families and genome groups. Although the DarkHorse algorithm cannot, by itself, provide definitive proof of horizontal gene transfer, it is a flexible, powerful tool that can be combined with slower, more rigorous methods in situations where these other methods could not otherwise be applied.
水平基因转移(HGT)过程被认为在细菌和古生菌中广泛存在,但关于其在完整微生物基因组中的发生情况,可获得的比较数据很少。由于计算需求过高,以前基于系统发育证据收集高质量、自动化的HGT预测数据对于大量基因组来说是不切实际的。DarkHorse是一种最近描述的用于在全基因组范围内发现系统发育非典型基因的统计方法,它通过谱系概率指数(LPI)排名分数提供了解决这个问题的方法。LPI分数反向反映测试氨基酸序列与其最接近的可用数据库匹配之间的系统发育距离。LPI分数低的蛋白质是水平基因转移的良好候选者;分数高的则不是。
DarkHorse算法已应用于955个微生物基因组序列,结果整理成一个可通过网络搜索的关系数据库,称为DarkHorse HGT候选资源库(http://darkhorse.ucsd.edu)。用户可以选择单个基因组或基因组组,通过LPI分数进行筛选,通过描述性注释或氨基酸序列相似性搜索蛋白质功能,或选择其基础编码序列中具有异常G+C组成的蛋白质。搜索引擎会报告匹配伙伴以及查询序列的LPI分数,从而有机会探索潜在的HGT供体序列在其自身基因组中是系统发育典型还是非典型的。这些信息可用于预测是否有足够的信息使用潜在的供体序列构建一个有充分支持的系统发育树。
DarkHorse HGT候选数据库提供了一套强大、灵活的工具,用于识别系统发育非典型蛋白质,使研究人员能够探索单个基因组中的单个HGT事件以及蛋白质家族和基因组组之间的大规模HGT模式。虽然DarkHorse算法本身不能提供水平基因转移的确凿证据,但它是一个灵活、强大的工具,可以在其他方法无法应用的情况下与较慢、更严格的方法结合使用。