Delport Wayne, Scheffler Konrad, Seoighe Cathal
Institute of Infectious Disease and Molecular Medicine, University of Cape Town, Rondebosch, Cape Town, South Africa.
PLoS Pathog. 2008 Dec;4(12):e1000242. doi: 10.1371/journal.ppat.1000242. Epub 2008 Dec 19.
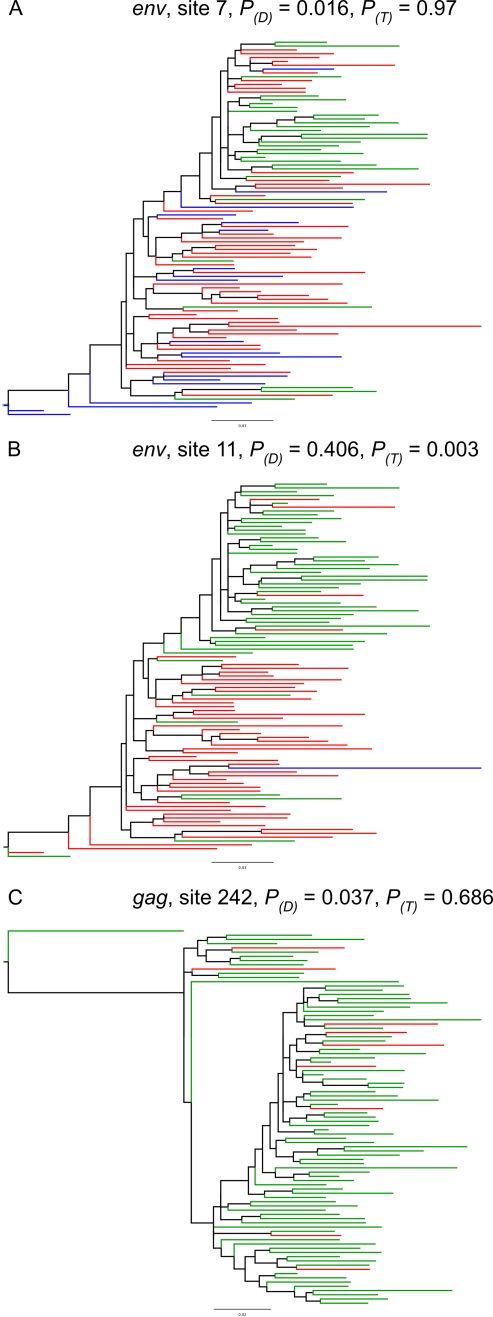
Host immune responses against infectious pathogens exert strong selective pressures favouring the emergence of escape mutations that prevent immune recognition. Escape mutations within or flanking functionally conserved epitopes can occur at a significant cost to the pathogen in terms of its ability to replicate effectively. Such mutations come under selective pressure to revert to the wild type in hosts that do not mount an immune response against the epitope. Amino acid positions exhibiting this pattern of escape and reversion are of interest because they tend to coincide with immune responses that control pathogen replication effectively. We have used a probabilistic model of protein coding sequence evolution to detect sites in HIV-1 exhibiting a pattern of rapid escape and reversion. Our model is designed to detect sites that toggle between a wild type amino acid, which is susceptible to a specific immune response, and amino acids with lower replicative fitness that evade immune recognition. Through simulation, we show that this model has significantly greater power to detect selection involving immune escape and reversion than standard models of diversifying selection, which are sensitive to an overall increased rate of non-synonymous substitution. Applied to alignments of HIV-1 protein coding sequences, the model of immune escape and reversion detects a significantly greater number of adaptively evolving sites in env and nef. In all genes tested, the model provides a significantly better description of adaptively evolving sites than standard models of diversifying selection. Several of the sites detected are corroborated by association between Human Leukocyte Antigen (HLA) and viral sequence polymorphisms. Overall, there is evidence for a large number of sites in HIV-1 evolving under strong selective pressure, but exhibiting low sequence diversity. A phylogenetic model designed to detect rapid toggling between wild type and escape amino acids identifies a larger number of adaptively evolving sites in HIV-1, and can in some cases correctly identify the amino acid that is susceptible to the immune response.
宿主针对传染性病原体的免疫反应会施加强大的选择压力,有利于出现阻止免疫识别的逃逸突变。功能保守表位内部或侧翼的逃逸突变可能会使病原体在有效复制能力方面付出巨大代价。在不对该表位产生免疫反应的宿主中,此类突变会受到选择压力,倾向于回复为野生型。呈现这种逃逸和回复模式的氨基酸位点很值得关注,因为它们往往与有效控制病原体复制的免疫反应相吻合。我们使用了一种蛋白质编码序列进化的概率模型来检测HIV-1中呈现快速逃逸和回复模式的位点。我们的模型旨在检测在易受特定免疫反应影响的野生型氨基酸与逃避免疫识别但复制适应性较低的氨基酸之间切换的位点。通过模拟,我们表明该模型在检测涉及免疫逃逸和回复的选择方面,比多样化选择的标准模型具有显著更强的能力,后者对非同义替换总体速率的增加较为敏感。将免疫逃逸和回复模型应用于HIV-1蛋白质编码序列比对时,该模型在env和nef中检测到数量显著更多的适应性进化位点。在所有测试基因中,该模型对适应性进化位点的描述比多样化选择的标准模型要好得多。检测到的几个位点得到了人类白细胞抗原(HLA)与病毒序列多态性之间关联的证实。总体而言,有证据表明HIV-1中有大量位点在强大的选择压力下进化,但序列多样性较低。一个旨在检测野生型和逃逸氨基酸之间快速切换的系统发育模型在HIV-1中识别出了更多适应性进化位点,并且在某些情况下能够正确识别易受免疫反应影响的氨基酸。