Wang Hao, Straubinger Robert M, Aletta John M, Cao Jin, Duan Xiaotao, Yu Haoying, Qu Jun
Department of Pharmaceutical Sciences, University at Buffalo, State University of New York, Amherst, New York, USA.
J Am Soc Mass Spectrom. 2009 Mar;20(3):507-19. doi: 10.1016/j.jasms.2008.11.008. Epub 2008 Nov 21.
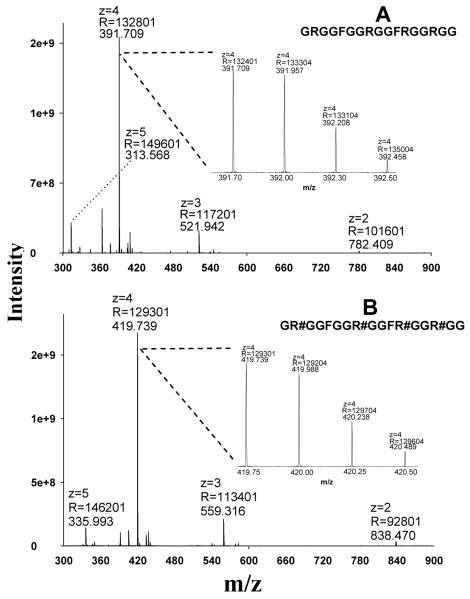
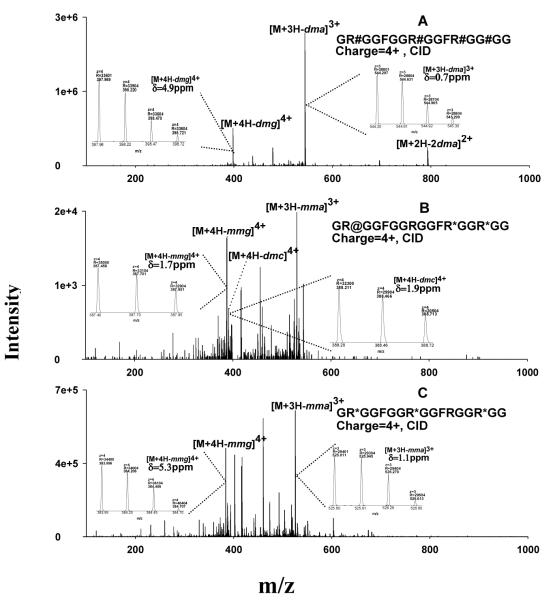
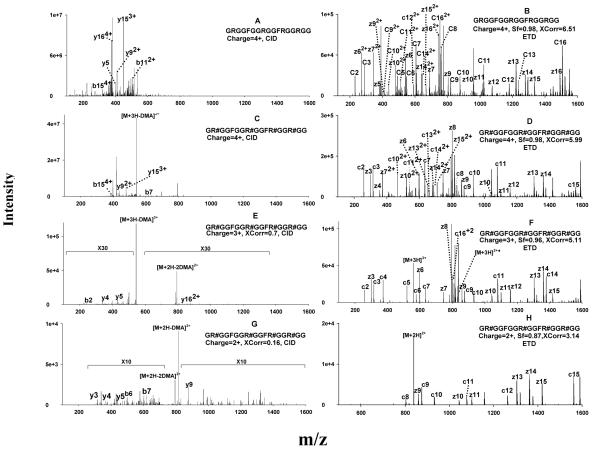
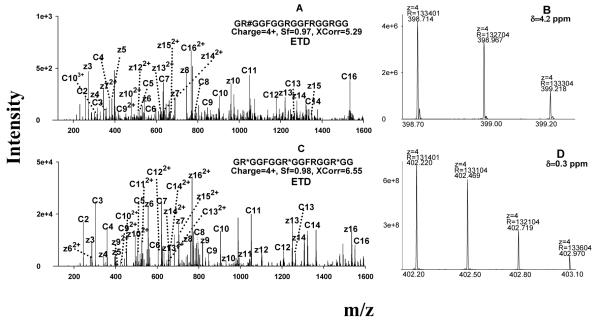
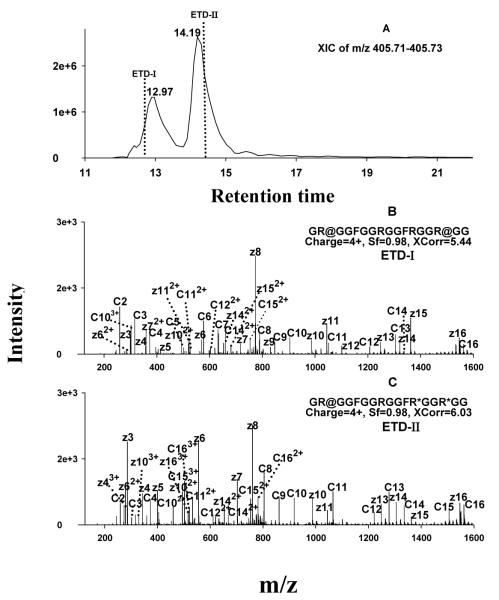
Protein arginine (Arg) methylation serves an important functional role in eucaryotic cells, and typically occurs in domains consisting of multiple Arg in close proximity. Localization of methylarginine (MA) within Arg-rich domains poses a challenge for mass spectrometry (MS)-based methods; the peptides are highly charged under electrospray ionization (ESI), which limits the number of sequence-informative products produced by collision induced dissociation (CID), and loss of the labile methylation moieties during CID precludes effective fragmentation of the peptide backbone. Here the fragmentation behavior of Arg-rich peptides was investigated comprehensively using electron-transfer dissociation (ETD) and CID for both methylated and unmodified glycine-/Arg-rich peptides (GAR), derived from residues 679-695 of human nucleolin, which contains methylation motifs that are widely-represented in biological systems. ETD produced abundant information for sequencing and MA localization, whereas CID failed to provide credible identification for any available charge state (z = 2-4). Nevertheless, CID produced characteristic neutral losses that can be employed to distinguish among different types of MA, as suggested by previous works and confirmed here with product ion scans of high accuracy/resolution by an LTQ/Orbitrap. To analyze MA-peptides in relatively complex mixtures, a method was developed that employs nano-LC coupled to alternating CID/ETD for peptide sequencing and MA localization/characterization, and an Orbitrap for accurate precursor measurement and relative quantification of MA-peptide stoichiometries. As proof of concept, GAR-peptides methylated in vitro by protein arginine N-methyltransferases PRMT1 and PRMT7 were analyzed. It was observed that PRMT1 generated a number of monomethylated (MMA) and asymmetric-dimethylated peptides, while PRMT7 produced predominantly MMA peptides and some symmetric-dimethylated peptides. This approach and the results may advance understanding of the actions of PRMTs and the functional significance of Arg methylation patterns.
蛋白质精氨酸(Arg)甲基化在真核细胞中发挥着重要的功能作用,通常发生在由多个相邻精氨酸组成的结构域中。甲基精氨酸(MA)在富含精氨酸的结构域中的定位对基于质谱(MS)的方法构成了挑战;这些肽在电喷雾电离(ESI)下带高电荷,这限制了碰撞诱导解离(CID)产生的序列信息产物的数量,并且CID过程中不稳定的甲基化部分的丢失妨碍了肽主链的有效断裂。在此,我们全面研究了富含精氨酸的肽的裂解行为,使用电子转移解离(ETD)和CID对来自人核仁素679 - 695位残基的甲基化和未修饰的富含甘氨酸/精氨酸的肽(GAR)进行分析,该肽段包含在生物系统中广泛存在的甲基化基序。ETD产生了丰富的测序和MA定位信息,而CID对于任何可用电荷态(z = 2 - 4)都未能提供可靠的鉴定。然而,正如先前工作所表明并在此通过LTQ/Orbitrap的高精度/分辨率产物离子扫描所证实的,CID产生了可用于区分不同类型MA的特征性中性丢失。为了分析相对复杂混合物中的MA - 肽,开发了一种方法,该方法采用纳升液相色谱与交替的CID/ETD联用进行肽测序和MA定位/表征,并使用Orbitrap进行MA - 肽化学计量的精确前体测量和相对定量。作为概念验证,分析了由蛋白质精氨酸N - 甲基转移酶PRMT1和PRMT7体外甲基化的GAR - 肽。观察到PRMT1产生了许多单甲基化(MMA)和不对称二甲基化肽,而PRMT7主要产生MMA肽和一些对称二甲基化肽。这种方法和结果可能会促进对PRMTs作用以及精氨酸甲基化模式功能意义的理解。