Paupe Vincent, Dassa Emmanuel P, Goncalves Sergio, Auchère Françoise, Lönn Maria, Holmgren Arne, Rustin Pierre
Inserm, U676, Hôpital Robert Debré, Bât. Ecran, Paris, France.
PLoS One. 2009;4(1):e4253. doi: 10.1371/journal.pone.0004253. Epub 2009 Jan 22.
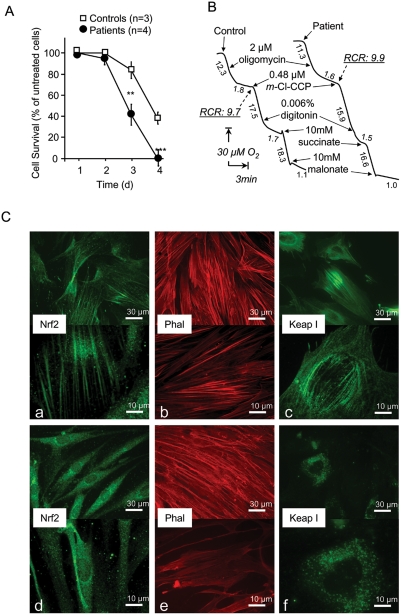
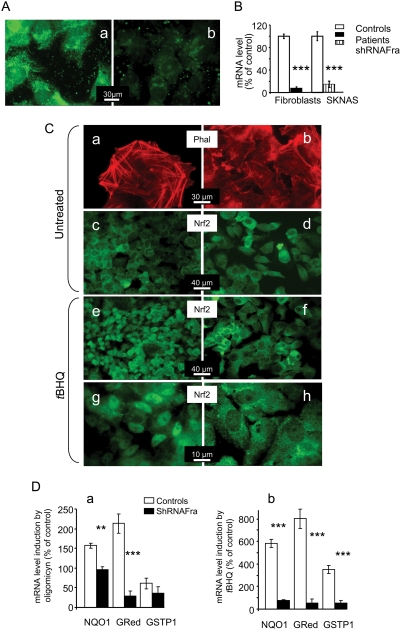
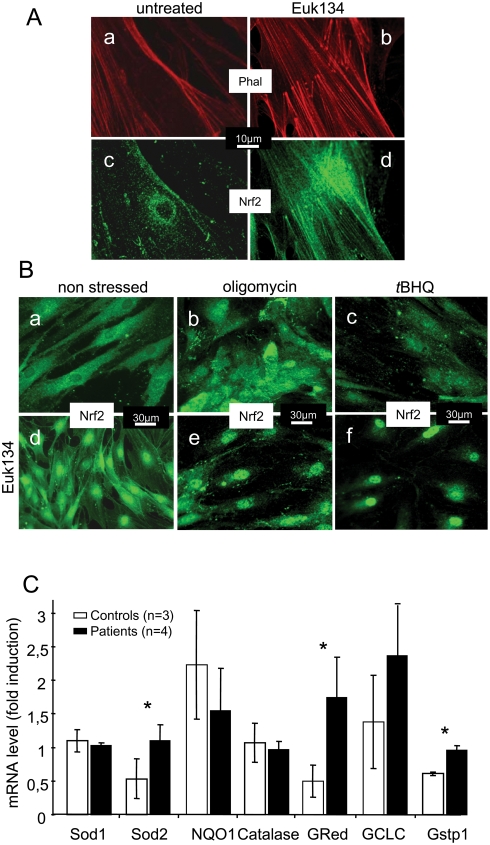
Friedreich ataxia originates from a decrease in mitochondrial frataxin, which causes the death of a subset of neurons. The biochemical hallmarks of the disease include low activity of the iron sulfur cluster-containing proteins (ISP) and impairment of antioxidant defense mechanisms that may play a major role in disease progression.
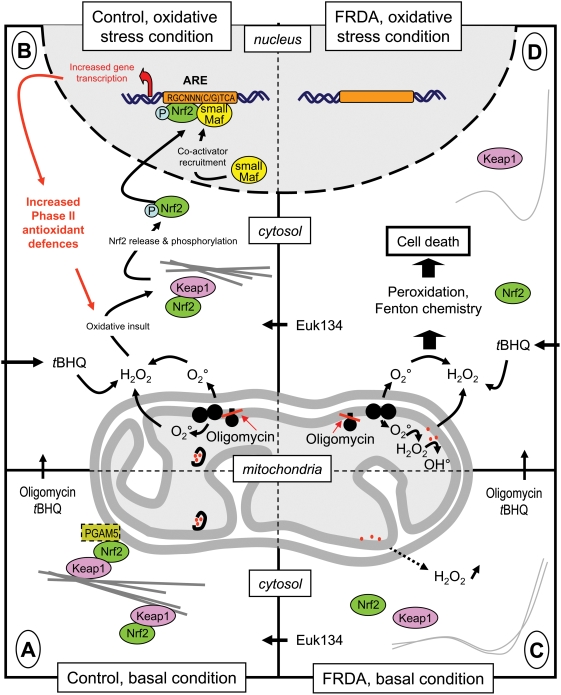
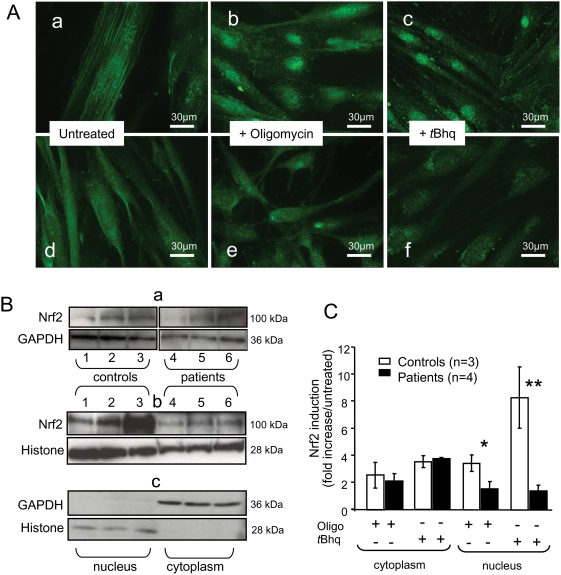
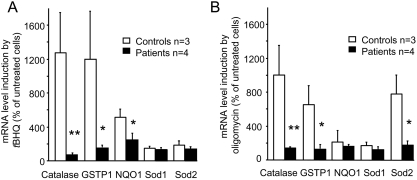
METHODOLOGY/PRINCIPAL FINDINGS: We thus investigated signaling pathways involved in antioxidant defense mechanisms. We showed that cultured fibroblasts from patients with Friedreich ataxia exhibited hypersensitivity to oxidative insults because of an impairment in the Nrf2 signaling pathway, which led to faulty induction of antioxidant enzymes. This impairment originated from previously reported actin remodeling by hydrogen peroxide.
CONCLUSIONS/SIGNIFICANCE: Thus, the defective machinery for ISP synthesis by causing mitochondrial iron dysmetabolism increases hydrogen peroxide production that accounts for the increased susceptibility to oxidative stress.
弗里德赖希共济失调源于线粒体铁硫蛋白减少,这会导致一部分神经元死亡。该疾病的生化特征包括含铁硫簇蛋白(ISP)活性降低以及抗氧化防御机制受损,而抗氧化防御机制受损可能在疾病进展中起主要作用。
方法/主要发现:因此,我们研究了参与抗氧化防御机制的信号通路。我们发现,弗里德赖希共济失调患者的培养成纤维细胞由于Nrf2信号通路受损而对氧化损伤表现出超敏反应,这导致抗氧化酶的诱导出现故障。这种损伤源于先前报道的过氧化氢引起的肌动蛋白重塑。
结论/意义:因此,由线粒体铁代谢异常导致的ISP合成缺陷机制会增加过氧化氢的产生,这解释了对氧化应激易感性增加的原因。