Cosentino Lagomarsino Marco, Sellerio Alessandro L, Heijning Philip D, Bassetti Bruno
Università degli Studi di Milano, Dip Fisica Milano, Italy.
Genome Biol. 2009;10(1):R12. doi: 10.1186/gb-2009-10-1-r12. Epub 2009 Jan 30.
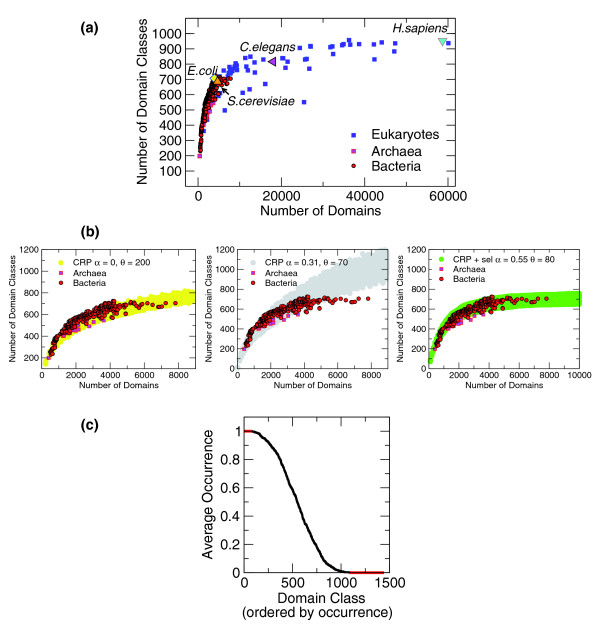
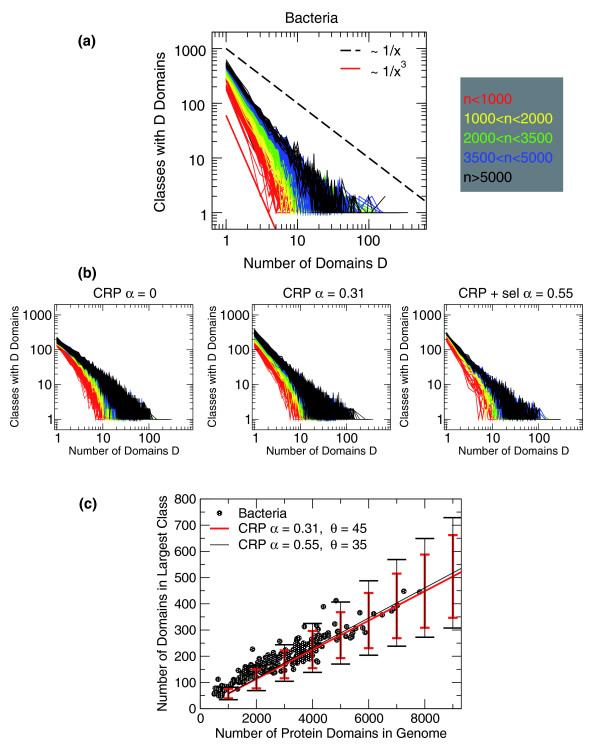
Protein domains can be used to study proteome evolution at a coarse scale. In particular, they are found on genomes with notable statistical distributions. It is known that the distribution of domains with a given topology follows a power law. We focus on a further aspect: these distributions, and the number of distinct topologies, follow collective trends, or scaling laws, depending on the total number of domains only, and not on genome-specific features.
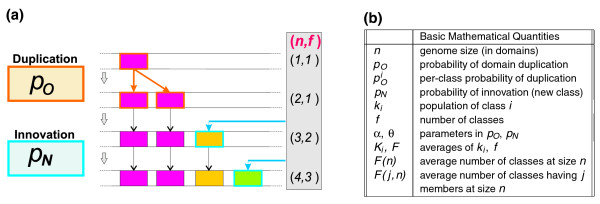
We present a stochastic duplication/innovation model, in the class of the so-called 'Chinese restaurant processes', that explains this observation with two universal parameters, representing a minimal number of domains and the relative weight of innovation to duplication. Furthermore, we study a model variant where new topologies are related to occurrence in genomic data, accounting for fold specificity.
Both models have general quantitative agreement with data from hundreds of genomes, which indicates that the domains of a genome are built with a combination of specificity and robust self-organizing phenomena. The latter are related to the basic evolutionary 'moves' of duplication and innovation, and give rise to the observed scaling laws, a priori of the specific evolutionary history of a genome. We interpret this as the concurrent effect of neutral and selective drives, which increase duplication and decrease innovation in larger and more complex genomes. The validity of our model would imply that the empirical observation of a small number of folds in nature may be a consequence of their evolution.
蛋白质结构域可用于在粗略尺度上研究蛋白质组的进化。特别是,它们在基因组上具有显著的统计分布。已知具有给定拓扑结构的结构域的分布遵循幂律。我们关注的是另一个方面:这些分布以及不同拓扑结构的数量遵循集体趋势或标度律,仅取决于结构域的总数,而不取决于基因组的特定特征。
我们提出了一种随机复制/创新模型,属于所谓的“中餐厅过程”类别,该模型用两个通用参数解释了这一观察结果,这两个参数分别代表结构域的最小数量以及创新与复制的相对权重。此外,我们研究了一种模型变体,其中新的拓扑结构与基因组数据中的出现情况相关,考虑了折叠特异性。
这两个模型在数量上都与来自数百个基因组的数据总体一致,这表明基因组的结构域是由特异性和强大的自组织现象共同构建而成的。后者与复制和创新这两个基本的进化“步骤”相关,并产生了观察到的标度律,这是先于基因组特定进化历史的。我们将此解释为中性和选择性驱动的共同作用,在更大、更复杂的基因组中,中性驱动增加复制,选择性驱动减少创新。我们模型的有效性意味着自然界中少数折叠的经验观察结果可能是其进化的结果。