Gene Expression Unit, European Molecular Biology Laboratory (EMBL), Meyerhofstr, Heidelberg, 69117, Germany.
Genome Biol. 2009 Feb 23;10(2):R23. doi: 10.1186/gb-2009-10-2-r23.
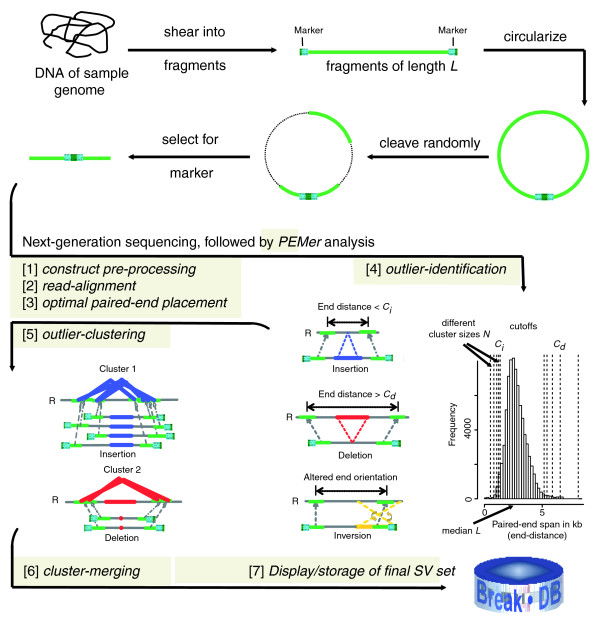
Personal-genomics endeavors, such as the 1000 Genomes project, are generating maps of genomic structural variants by analyzing ends of massively sequenced genome fragments. To process these we developed Paired-End Mapper (PEMer; http://sv.gersteinlab.org/pemer). This comprises an analysis pipeline, compatible with several next-generation sequencing platforms; simulation-based error models, yielding confidence-values for each structural variant; and a back-end database. The simulations demonstrated high structural variant reconstruction efficiency for PEMer's coverage-adjusted multi-cutoff scoring-strategy and showed its relative insensitivity to base-calling errors.
个人基因组学研究,如 1000 基因组计划,通过分析大量测序的基因组片段的末端来生成基因组结构变异图谱。为了处理这些数据,我们开发了 Paired-End Mapper(PEMer;http://sv.gersteinlab.org/pemer)。它包括一个分析管道,兼容几种下一代测序平台;基于模拟的错误模型,为每个结构变异产生置信值;以及后端数据库。模拟表明,PEMer 的覆盖调整多切割评分策略具有很高的结构变异重建效率,并且对碱基呼叫错误相对不敏感。