Sayed Samir, Langdon David R, Odili Stella, Chen Pan, Buettger Carol, Schiffman Alisa B, Suchi Mariko, Taub Rebecca, Grimsby Joseph, Matschinsky Franz M, Stanley Charles A
Clinical Translational Research Center, Children's Hospital of Philadelphia, Philadelphia, Pennsylvania, USA.
Diabetes. 2009 Jun;58(6):1419-27. doi: 10.2337/db08-1792. Epub 2009 Mar 31.
Heterozygous activating mutations of glucokinase have been reported to cause hypoglycemia attributable to hyperinsulinism in a limited number of families. We report three children with de novo glucokinase hyperinsulinism mutations who displayed a spectrum of clinical phenotypes corresponding to marked differences in enzyme kinetics.
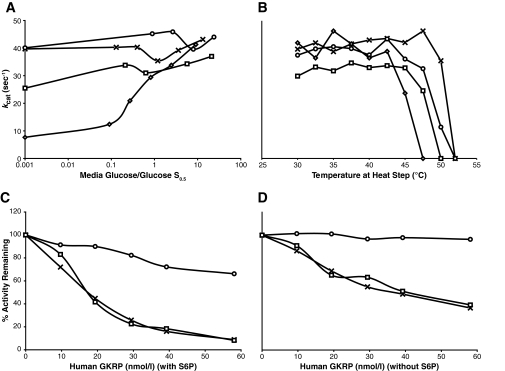
Mutations were directly sequenced, and mutants were expressed as glutathionyl S-transferase-glucokinase fusion proteins. Kinetic analysis of the enzymes included determinations of stability, activity index, the response to glucokinase activator drug, and the effect of glucokinase regulatory protein.
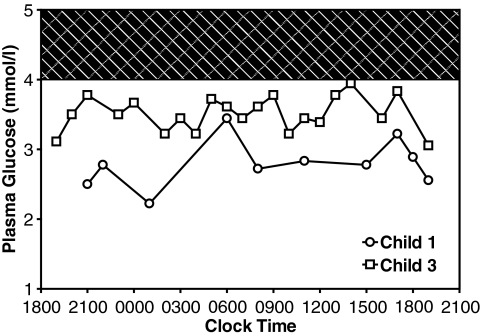
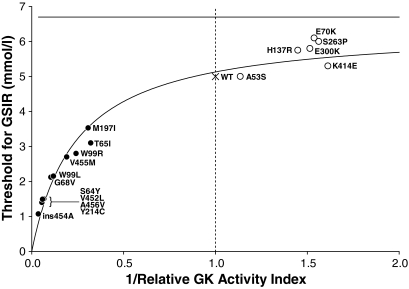
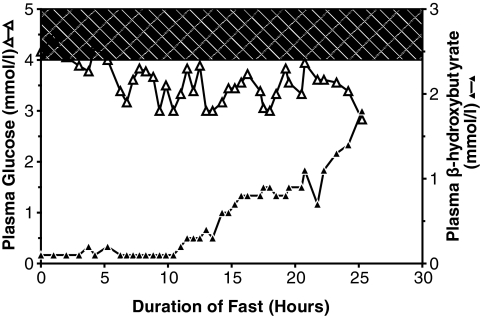
Child 1 had an ins454A mutation, child 2 a W99L mutation, and child 3 an M197I mutation. Diazoxide treatment was effective in child 3 but ineffective in child 1 and only partially effective in child 2. Expression of the mutant glucokinase ins454A, W99L, and M197I enzymes revealed a continuum of high relative activity indexes in the three children (26, 8.9, and 3.1, respectively; wild type = 1.0). Allosteric responses to inhibition by glucokinase regulatory protein and activation by the drug RO0281675 were impaired by the ins454A but unaffected by the M197I mutation. Estimated thresholds for glucose-stimulated insulin release were more severely reduced by the ins454A than the M197I mutation and intermediate in the W99L mutation (1.1, 3.5, and 2.2 mmol/l, respectively; wild type = 5.0 mmol/l).
These results confirm the potency of glucokinase as the pancreatic beta-cell glucose sensor, and they demonstrate that responsiveness to diazoxide varies with genotype in glucokinase hyperinsulinism resulting in hypoglycemia, which can be more difficult to control than previously believed.
据报道,在少数家庭中,葡萄糖激酶的杂合激活突变可导致因高胰岛素血症引起的低血糖。我们报告了三名患有新发葡萄糖激酶高胰岛素血症突变的儿童,他们表现出一系列临床表型,与酶动力学的显著差异相对应。
对突变进行直接测序,并将突变体表达为谷胱甘肽S-转移酶-葡萄糖激酶融合蛋白。酶的动力学分析包括稳定性测定、活性指数测定、对葡萄糖激酶激活剂药物的反应以及葡萄糖激酶调节蛋白的作用。
患儿1有ins454A突变,患儿2有W99L突变,患儿3有M197I突变。二氮嗪治疗对患儿3有效,但对患儿1无效,对患儿2仅部分有效。突变型葡萄糖激酶ins454A、W99L和M197I酶的表达显示,三名儿童的相对活性指数呈连续变化(分别为26、8.9和3.1;野生型 = 1.0)。ins454A突变损害了对葡萄糖激酶调节蛋白抑制和药物RO0281675激活的变构反应,但M197I突变未受影响。ins454A突变比M197I突变更严重地降低了葡萄糖刺激的胰岛素释放的估计阈值,W99L突变的阈值处于中间水平(分别为1.1、3.5和2.2 mmol/L;野生型 = 5.0 mmol/L)。
这些结果证实了葡萄糖激酶作为胰腺β细胞葡萄糖传感器的作用,并且表明在导致低血糖的葡萄糖激酶高胰岛素血症中,对二氮嗪的反应性因基因型而异——这可能比之前认为的更难控制。