Allen Jonathan E, Gardner Shea N, Vitalis Elizabeth A, Slezak Tom R
Lawrence Livermore National Laboratory, Livermore, CA, 94551, USA.
BMC Microbiol. 2009 Apr 22;9:77. doi: 10.1186/1471-2180-9-77.
Finding the amino acid mutations that affect the severity of influenza infections remains an open and challenging problem. Of special interest is better understanding how current circulating influenza strains could evolve into a new pandemic strain. Influenza proteomes from distinct viral phenotype classes were searched for class specific amino acid mutations conserved in past pandemics, using reverse engineered linear classifiers.
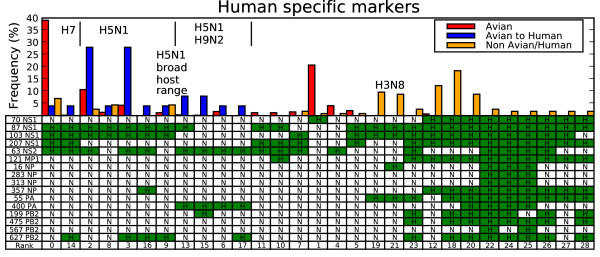
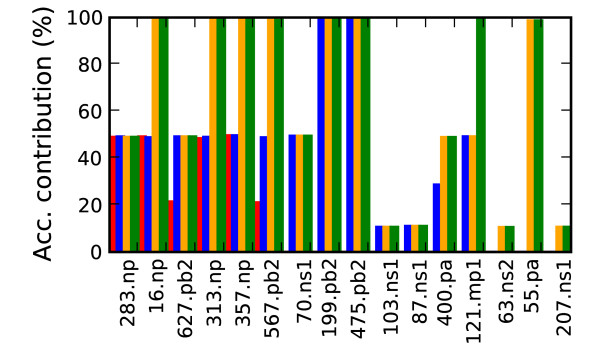
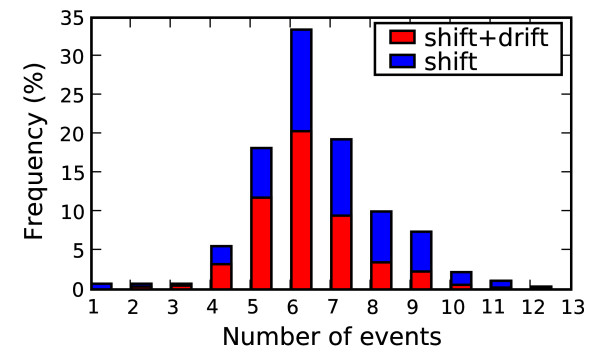
Thirty-four amino acid markers associated with host specificity and high mortality rate were found. Some markers had little impact on distinguishing the functional classes by themselves, however in combination with other mutations they improved class prediction. Pairwise combinations of influenza genomes were checked for reassortment and mutation events needed to acquire the pandemic conserved markers. Evolutionary pathways involving H1N1 human and swine strains mixed with avian strains show the potential to acquire the pandemic markers with a double reassortment and one or two amino acid mutations.
The small mutation combinations found at multiple protein positions associated with viral phenotype indicate that surveillance tools could monitor genetic variation beyond single point mutations to track influenza strains. Finding that certain strain combinations have the potential to acquire pandemic conserved markers through a limited number of reassortment and mutation events illustrates the potential for reassortment and mutation events to lead to new circulating influenza strains.
寻找影响流感感染严重程度的氨基酸突变仍然是一个尚未解决且具有挑战性的问题。特别令人感兴趣的是更好地理解当前正在传播的流感毒株如何演变成新的大流行毒株。利用反向工程线性分类器,在不同病毒表型类别的流感蛋白质组中搜索过去大流行中保守的类别特异性氨基酸突变。
发现了34个与宿主特异性和高死亡率相关的氨基酸标记。一些标记本身对区分功能类别影响不大,但与其他突变结合时可改善类别预测。检查了流感基因组的成对组合,以寻找获得大流行保守标记所需的重配和突变事件。涉及H1N1人类和猪毒株与禽毒株混合的进化途径显示,通过双重重配和一两个氨基酸突变获得大流行标记的可能性。
在与病毒表型相关的多个蛋白质位置发现的小突变组合表明,监测工具可以监测单点突变以外的遗传变异,以追踪流感毒株。发现某些毒株组合有可能通过有限数量的重配和突变事件获得大流行保守标记,这说明了重配和突变事件导致新的正在传播的流感毒株出现的可能性。