Friedrich Miescher Laboratory of the Max Planck Society, and Max Planck Institute for Developmental Biology, Tübingen, Germany.
Bioinformatics. 2009 Aug 15;25(16):2126-33. doi: 10.1093/bioinformatics/btp278. Epub 2009 Apr 23.
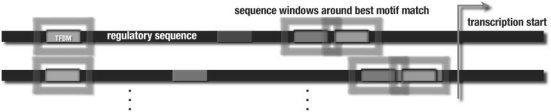
Understanding transcriptional regulation is one of the main challenges in computational biology. An important problem is the identification of transcription factor (TF) binding sites in promoter regions of potential TF target genes. It is typically approached by position weight matrix-based motif identification algorithms using Gibbs sampling, or heuristics to extend seed oligos. Such algorithms succeed in identifying single, relatively well-conserved binding sites, but tend to fail when it comes to the identification of combinations of several degenerate binding sites, as those often found in cis-regulatory modules.
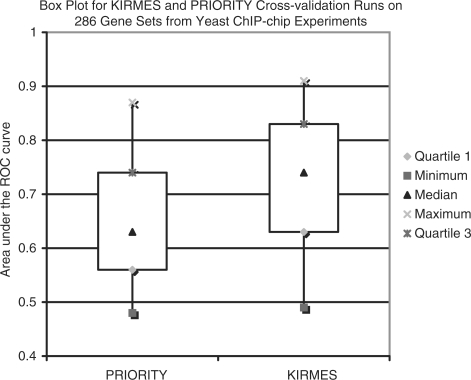
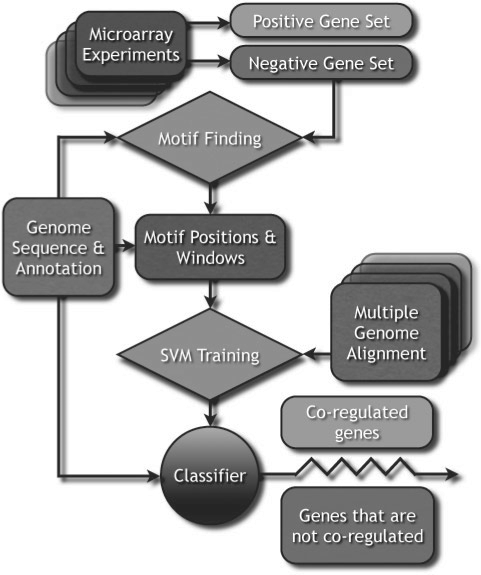
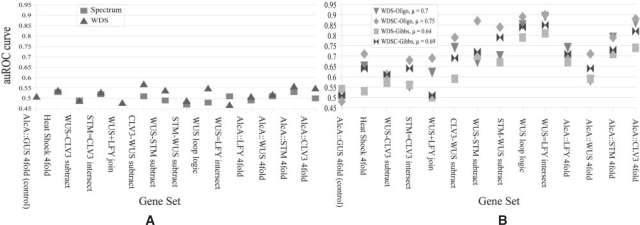
We propose a new algorithm that combines the benefits of existing motif finding with the ones of support vector machines (SVMs) to find degenerate motifs in order to improve the modeling of regulatory modules. In experiments on microarray data from Arabidopsis thaliana, we were able to show that the newly developed strategy significantly improves the recognition of TF targets.
The python source code (open source-licensed under GPL), the data for the experiments and a Galaxy-based web service are available at http://www.fml.mpg.de/raetsch/suppl/kirmes/.
理解转录调控是计算生物学中的主要挑战之一。一个重要的问题是识别潜在转录因子(TF)靶基因启动子区域中的 TF 结合位点。通常通过基于位置权重矩阵的 motif 识别算法使用 Gibbs 采样或启发式方法来扩展种子寡核苷酸来解决此问题。这些算法成功地识别了单个相对保守的结合位点,但在识别几个简并结合位点的组合时往往会失败,因为这些组合通常存在于顺式调控模块中。
我们提出了一种新算法,该算法结合了现有 motif 发现算法和支持向量机(SVM)的优势,以找到简并 motif,从而改进调控模块的建模。在对拟南芥的 microarray 数据进行的实验中,我们能够证明新开发的策略显著提高了 TF 靶标的识别能力。
python 源代码(在 GPL 下开源)、实验数据以及基于 Galaxy 的网络服务可在 http://www.fml.mpg.de/raetsch/suppl/kirmes/ 上获得。