Koo Qi Ying, Khan Asif M, Jung Keun-Ok, Ramdas Shweta, Miotto Olivo, Tan Tin Wee, Brusic Vladimir, Salmon Jerome, August J Thomas
Department of Biochemistry, Yong Loo Lin School of Medicine, National University of Singapore, Singapore, Singapore.
PLoS One. 2009;4(4):e5352. doi: 10.1371/journal.pone.0005352. Epub 2009 Apr 29.
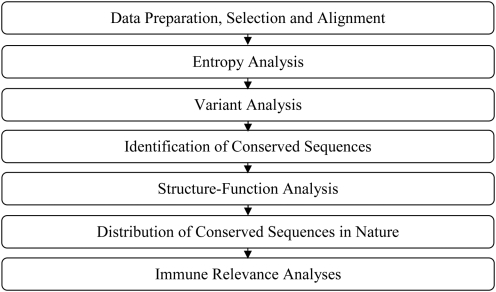
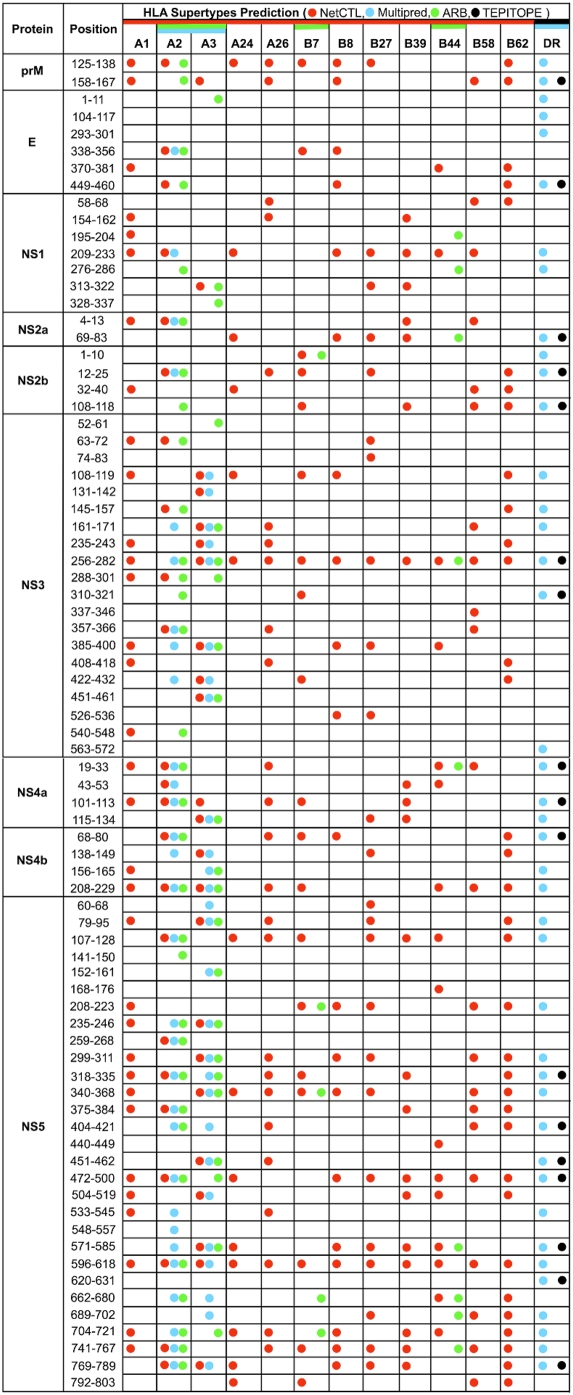
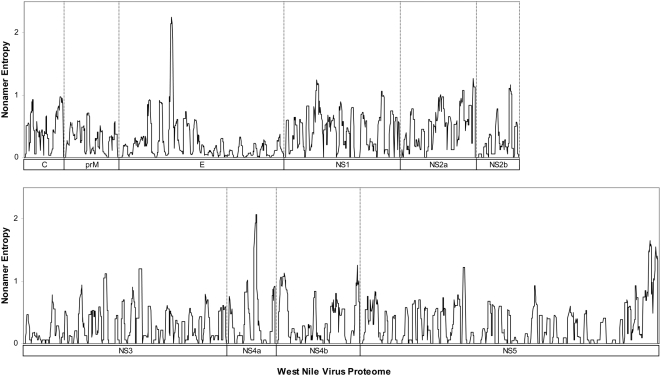
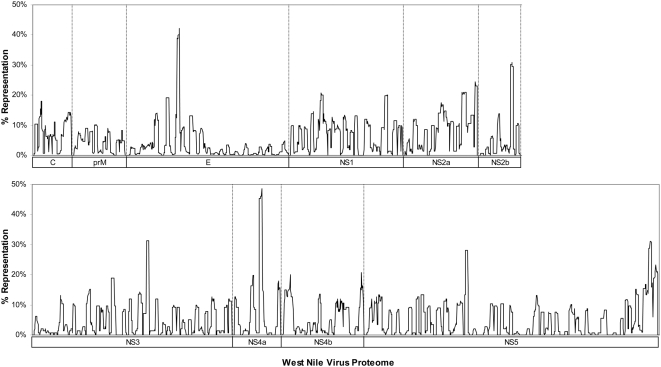
West Nile virus (WNV) has emerged globally as an increasingly important pathogen for humans and domestic animals. Studies of the evolutionary diversity of the virus over its known history will help to elucidate conserved sites, and characterize their correspondence to other pathogens and their relevance to the immune system. We describe a large-scale analysis of the entire WNV proteome, aimed at identifying and characterizing evolutionarily conserved amino acid sequences. This study, which used 2,746 WNV protein sequences collected from the NCBI GenPept database, focused on analysis of peptides of length 9 amino acids or more, which are immunologically relevant as potential T-cell epitopes. Entropy-based analysis of the diversity of WNV sequences, revealed the presence of numerous evolutionarily stable nonamer positions across the proteome (entropy value of < or = 1). The representation (frequency) of nonamers variant to the predominant peptide at these stable positions was, generally, low (< or = 10% of the WNV sequences analyzed). Eighty-eight fragments of length 9-29 amino acids, representing approximately 34% of the WNV polyprotein length, were identified to be identical and evolutionarily stable in all analyzed WNV sequences. Of the 88 completely conserved sequences, 67 are also present in other flaviviruses, and several have been associated with the functional and structural properties of viral proteins. Immunoinformatic analysis revealed that the majority (78/88) of conserved sequences are potentially immunogenic, while 44 contained experimentally confirmed human T-cell epitopes. This study identified a comprehensive catalogue of completely conserved WNV sequences, many of which are shared by other flaviviruses, and majority are potential epitopes. The complete conservation of these immunologically relevant sequences through the entire recorded WNV history suggests they will be valuable as components of peptide-specific vaccines or other therapeutic applications, for sequence-specific diagnosis of a wide-range of Flavivirus infections, and for studies of homologous sequences among other flaviviruses.
西尼罗河病毒(WNV)已在全球范围内成为对人类和家畜越来越重要的病原体。对该病毒已知历史上的进化多样性进行研究,将有助于阐明保守位点,并描述它们与其他病原体的对应关系及其与免疫系统的相关性。我们描述了对整个WNV蛋白质组的大规模分析,旨在识别和表征进化上保守的氨基酸序列。这项研究使用了从NCBI GenPept数据库收集的2746个WNV蛋白质序列,重点分析长度为9个氨基酸或更长的肽段,这些肽段作为潜在的T细胞表位具有免疫相关性。基于熵的WNV序列多样性分析揭示了整个蛋白质组中存在许多进化上稳定的九聚体位置(熵值≤1)。在这些稳定位置上与主要肽段不同的九聚体的代表性(频率)通常较低(≤所分析的WNV序列的10%)。在所有分析的WNV序列中,鉴定出88个长度为9 - 29个氨基酸的片段,约占WNV多蛋白长度的34%,它们是相同且进化上稳定的。在这88个完全保守的序列中,67个也存在于其他黄病毒中,并且有几个与病毒蛋白的功能和结构特性有关。免疫信息学分析表明,大多数(78/88)保守序列具有潜在免疫原性,而44个包含经实验证实的人类T细胞表位。这项研究确定了一个完整的WNV完全保守序列目录,其中许多序列为其他黄病毒所共有,并且大多数是潜在表位。这些免疫相关序列在整个记录的WNV历史中完全保守,这表明它们作为肽特异性疫苗或其他治疗应用的成分、用于多种黄病毒感染的序列特异性诊断以及用于研究其他黄病毒中的同源序列将具有重要价值。