Department of Biochemistry, Yong Loo Lin School of Medicine, National University of Singapore, Singapore.
PLoS Negl Trop Dis. 2008 Aug 13;2(8):e272. doi: 10.1371/journal.pntd.0000272.
Genetic variation and rapid evolution are hallmarks of RNA viruses, the result of high mutation rates in RNA replication and selection of mutants that enhance viral adaptation, including the escape from host immune responses. Variability is uneven across the genome because mutations resulting in a deleterious effect on viral fitness are restricted. RNA viruses are thus marked by protein sites permissive to multiple mutations and sites critical to viral structure-function that are evolutionarily robust and highly conserved. Identification and characterization of the historical dynamics of the conserved sites have relevance to multiple applications, including potential targets for diagnosis, and prophylactic and therapeutic purposes.
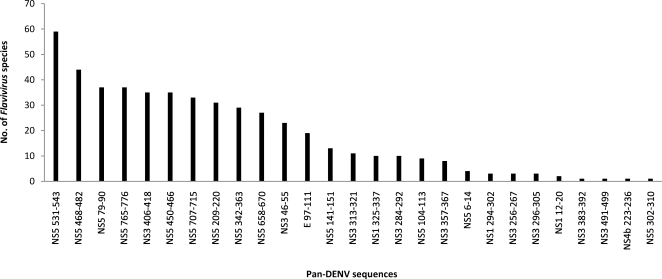
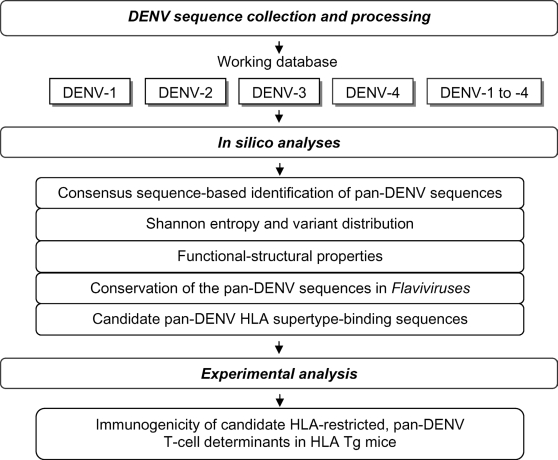
METHODOLOGY/PRINCIPAL FINDINGS: We describe a large-scale identification and analysis of evolutionarily highly conserved amino acid sequences of the entire dengue virus (DENV) proteome, with a focus on sequences of 9 amino acids or more, and thus immune-relevant as potential T-cell determinants. DENV protein sequence data were collected from the NCBI Entrez protein database in 2005 (9,512 sequences) and again in 2007 (12,404 sequences). Forty-four (44) sequences (pan-DENV sequences), mainly those of nonstructural proteins and representing approximately 15% of the DENV polyprotein length, were identical in 80% or more of all recorded DENV sequences. Of these 44 sequences, 34 ( approximately 77%) were present in >or=95% of sequences of each DENV type, and 27 ( approximately 61%) were conserved in other Flaviviruses. The frequencies of variants of the pan-DENV sequences were low (0 to approximately 5%), as compared to variant frequencies of approximately 60 to approximately 85% in the non pan-DENV sequence regions. We further showed that the majority of the conserved sequences were immunologically relevant: 34 contained numerous predicted human leukocyte antigen (HLA) supertype-restricted peptide sequences, and 26 contained T-cell determinants identified by studies with HLA-transgenic mice and/or reported to be immunogenic in humans.
CONCLUSIONS/SIGNIFICANCE: Forty-four (44) pan-DENV sequences of at least 9 amino acids were highly conserved and identical in 80% or more of all recorded DENV sequences, and the majority were found to be immune-relevant by their correspondence to known or putative HLA-restricted T-cell determinants. The conservation of these sequences through the entire recorded DENV genetic history supports their possible value for diagnosis, prophylactic and/or therapeutic applications. The combination of bioinformatics and experimental approaches applied herein provides a framework for large-scale and systematic analysis of conserved and variable sequences of other pathogens, in particular, for rapidly mutating viruses, such as influenza A virus and HIV.
遗传变异和快速进化是 RNA 病毒的特征,这是由于 RNA 复制过程中的高突变率以及选择增强病毒适应性的突变体的结果,包括逃避宿主免疫反应。基因组的变异性不均匀,因为导致病毒适应性降低的突变受到限制。因此,RNA 病毒的蛋白质位点允许多种突变,而对病毒结构和功能至关重要的位点则具有进化稳健性和高度保守性。鉴定和描述保守位点的历史动态与多种应用相关,包括诊断、预防和治疗目的的潜在目标。
方法/主要发现:我们描述了一种大规模识别和分析整个登革热病毒 (DENV) 蛋白质组中进化上高度保守的氨基酸序列的方法,重点是 9 个或更多氨基酸的序列,因此作为潜在的 T 细胞决定簇具有免疫相关性。DENV 蛋白质序列数据于 2005 年从 NCBI Entrez 蛋白质数据库中收集(9512 个序列),并于 2007 年再次收集(12404 个序列)。44 个(泛 DENV 序列),主要是非结构蛋白序列,代表 DENV 多蛋白长度的约 15%,在 80%或更多记录的 DENV 序列中是相同的。在这些 44 个序列中,34 个(约 77%)存在于每种 DENV 型的 95%以上的序列中,27 个(约 61%)在其他黄病毒中保守。泛 DENV 序列变体的频率较低(0 至约 5%),而非泛 DENV 序列区域的变体频率约为 60 至约 85%。我们进一步表明,大多数保守序列具有免疫相关性:34 个包含许多预测的人类白细胞抗原 (HLA) 超型限制性肽序列,26 个包含通过 HLA 转基因小鼠研究鉴定的 T 细胞决定簇,或报告在人类中具有免疫原性。
结论/意义:至少 9 个氨基酸的 44 个泛 DENV 序列高度保守,在 80%或更多记录的 DENV 序列中是相同的,并且通过与已知或假定的 HLA 限制性 T 细胞决定簇相对应,大多数被发现具有免疫相关性。这些序列在整个记录的 DENV 遗传历史中保持保守,支持它们在诊断、预防和/或治疗应用中的潜在价值。本文应用的生物信息学和实验方法的组合为大规模和系统地分析其他病原体的保守和可变序列提供了一个框架,特别是对于快速突变的病毒,如流感病毒和 HIV。