Wegdam Wouter, Moerland Perry D, Buist Marrije R, van Themaat Emiel Ver Loren, Bleijlevens Boris, Hoefsloot Huub Cj, de Koster Chris G, Aerts Johannes Mfg
Department of Gynaecologic Oncology, Academic Medical Center, University of Amsterdam, Amsterdam, the Netherlands.
Bioinformatics Laboratory, Department of Clinical Epidemiology, Biostatistics and Bioinformatics, Academic Medical Center, University of Amsterdam, Amsterdam, the Netherlands.
Proteome Sci. 2009 May 14;7:19. doi: 10.1186/1477-5956-7-19.
Mass spectrometry is increasingly being used to discover proteins or protein profiles associated with disease. Experimental design of mass-spectrometry studies has come under close scrutiny and the importance of strict protocols for sample collection is now understood. However, the question of how best to process the large quantities of data generated is still unanswered. Main challenges for the analysis are the choice of proper pre-processing and classification methods. While these two issues have been investigated in isolation, we propose to use the classification of patient samples as a clinically relevant benchmark for the evaluation of pre-processing methods.
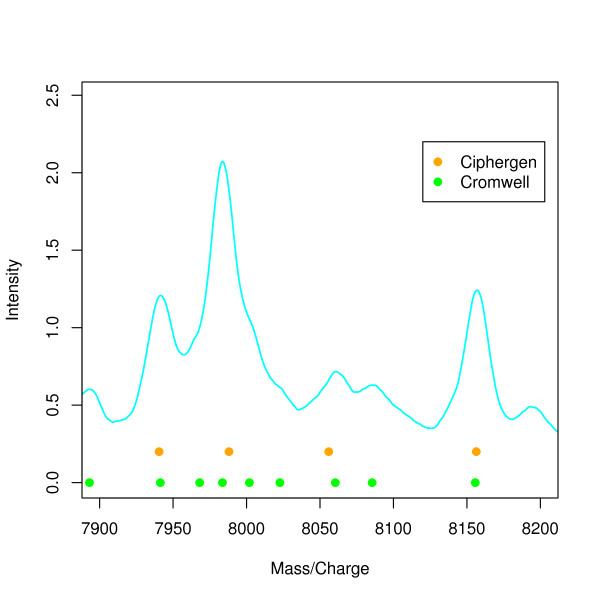
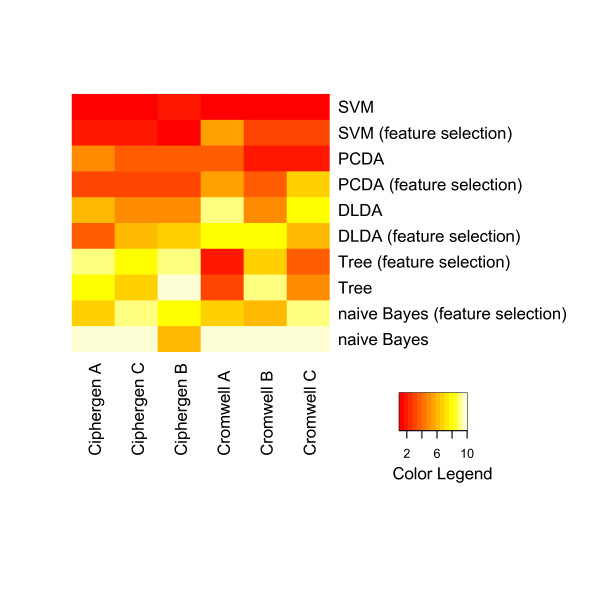
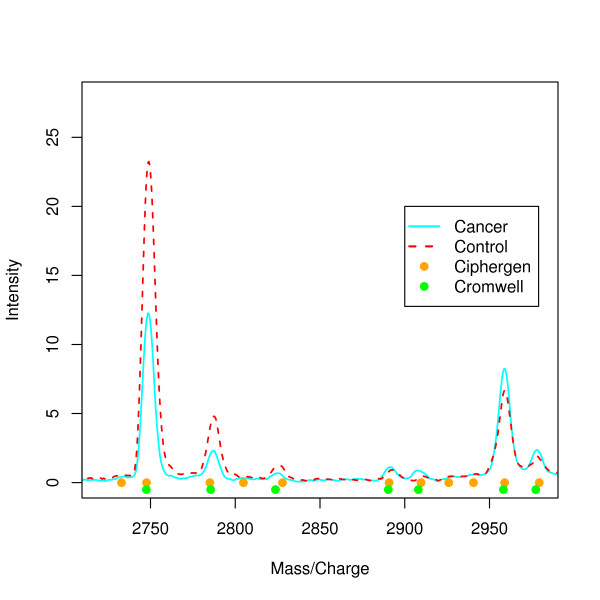
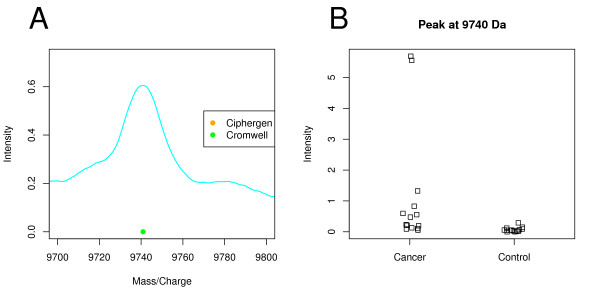
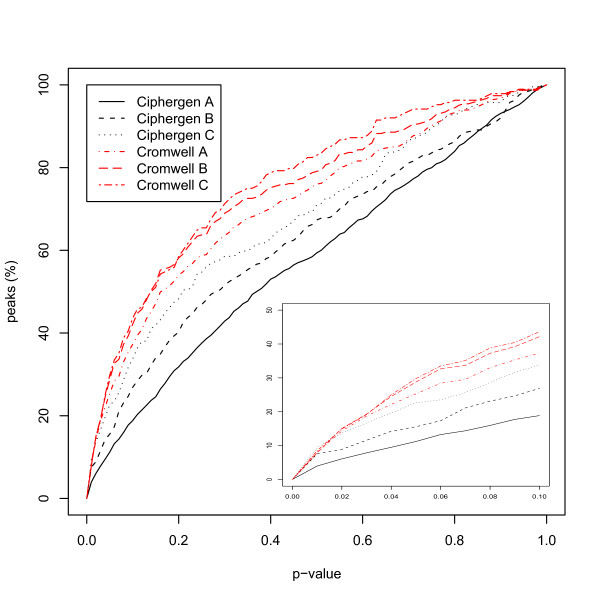
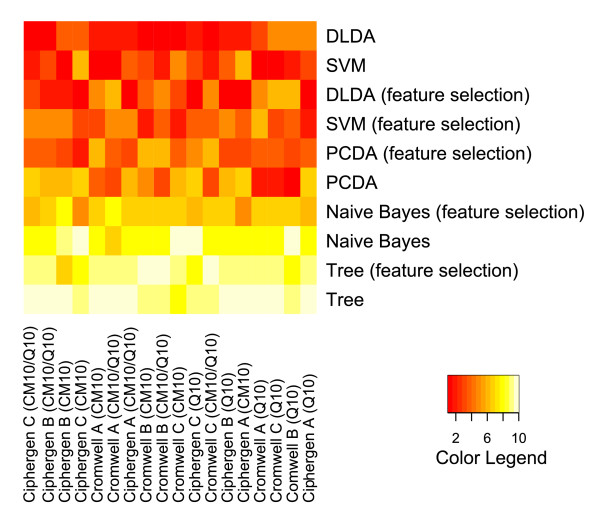
Two in-house generated clinical SELDI-TOF MS datasets are used in this study as an example of high throughput mass-spectrometry data. We perform a systematic comparison of two commonly used pre-processing methods as implemented in Ciphergen ProteinChip Software and in the Cromwell package. With respect to reproducibility, Ciphergen and Cromwell pre-processing are largely comparable. We find that the overlap between peaks detected by either Ciphergen ProteinChip Software or Cromwell is large. This is especially the case for the more stringent peak detection settings. Moreover, similarity of the estimated intensities between matched peaks is high.We evaluate the pre-processing methods using five different classification methods. Classification is done in a double cross-validation protocol using repeated random sampling to obtain an unbiased estimate of classification accuracy. No pre-processing method significantly outperforms the other for all peak detection settings evaluated.
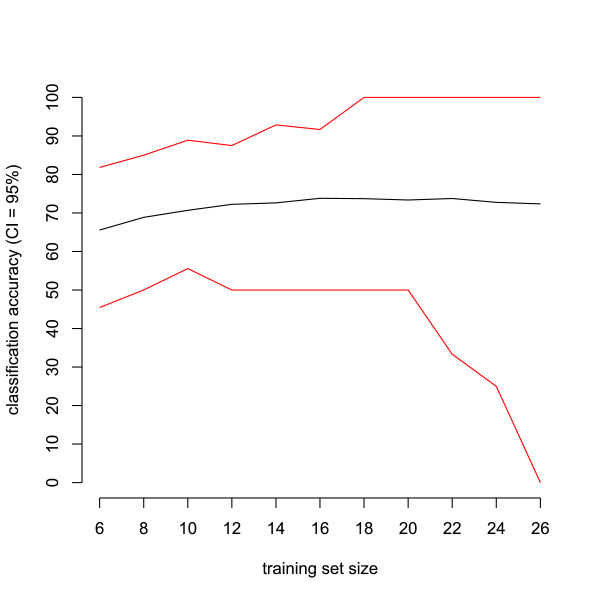
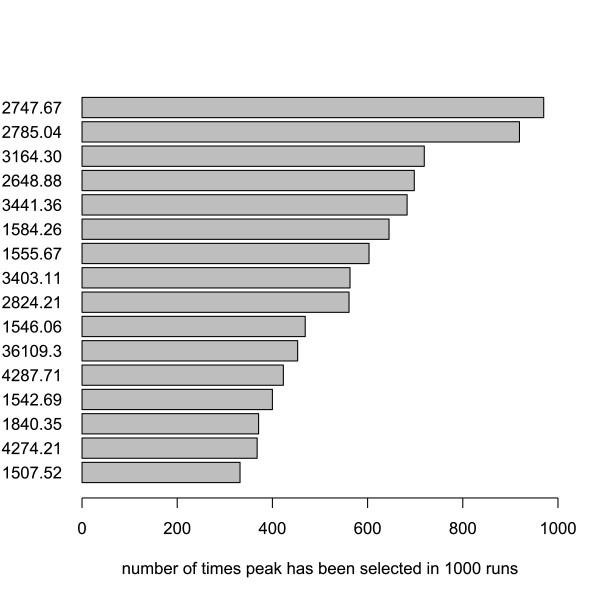
We use classification of patient samples as a clinically relevant benchmark for the evaluation of pre-processing methods. Both pre-processing methods lead to similar classification results on an ovarian cancer and a Gaucher disease dataset. However, the settings for pre-processing parameters lead to large differences in classification accuracy and are therefore of crucial importance. We advocate the evaluation over a range of parameter settings when comparing pre-processing methods. Our analysis also demonstrates that reliable classification results can be obtained with a combination of strict sample handling and a well-defined classification protocol on clinical samples.
质谱分析法越来越多地用于发现与疾病相关的蛋白质或蛋白质谱。质谱研究的实验设计受到了密切审查,现在人们已经认识到严格的样本采集方案的重要性。然而,如何最好地处理大量生成的数据这一问题仍未得到解答。分析的主要挑战在于选择合适的预处理和分类方法。虽然这两个问题已分别进行了研究,但我们建议将患者样本的分类作为评估预处理方法的临床相关基准。
本研究使用了两个内部生成的临床表面增强激光解吸电离飞行时间质谱(SELDI-TOF MS)数据集作为高通量质谱数据的示例。我们对Ciphergen蛋白质芯片软件和Cromwell软件包中实现的两种常用预处理方法进行了系统比较。在可重复性方面,Ciphergen和Cromwell预处理在很大程度上具有可比性。我们发现,Ciphergen蛋白质芯片软件或Cromwell检测到的峰之间的重叠很大。在更严格的峰检测设置下尤其如此。此外,匹配峰之间估计强度的相似度很高。我们使用五种不同的分类方法评估预处理方法。分类是在双重交叉验证方案中进行的,使用重复随机抽样来获得分类准确性的无偏估计。对于所有评估的峰检测设置,没有一种预处理方法明显优于其他方法。
我们将患者样本的分类作为评估预处理方法的临床相关基准。两种预处理方法在卵巢癌和戈谢病数据集上都产生了相似的分类结果。然而,预处理参数的设置导致分类准确性存在很大差异,因此至关重要。我们主张在比较预处理方法时对一系列参数设置进行评估。我们的分析还表明,通过对临床样本进行严格的样本处理和明确的分类方案相结合,可以获得可靠的分类结果。