Garber Manuel, Guttman Mitchell, Clamp Michele, Zody Michael C, Friedman Nir, Xie Xiaohui
Department of Biology, Broad Institute of MIT and Harvard, 7 Cambridge Center, MIT, Cambridge, MA 02142, USA.
Bioinformatics. 2009 Jun 15;25(12):i54-62. doi: 10.1093/bioinformatics/btp190.
Comparing the genomes from closely related species provides a powerful tool to identify functional elements in a reference genome. Many methods have been developed to identify conserved sequences across species; however, existing methods only model conservation as a decrease in the rate of mutation and have ignored selection acting on the pattern of mutations.
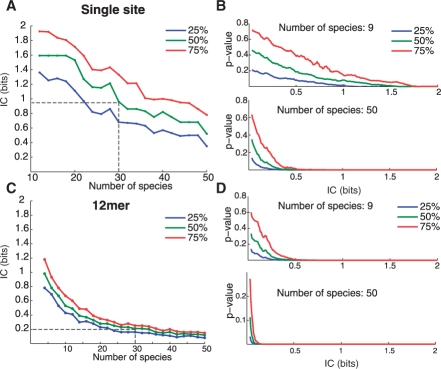
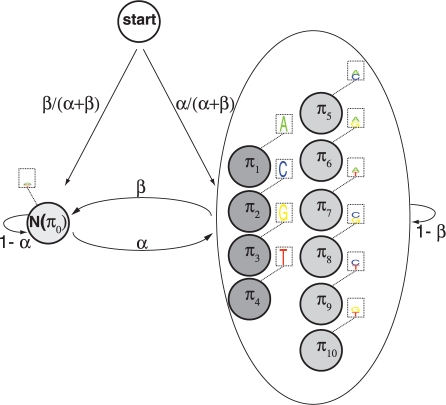
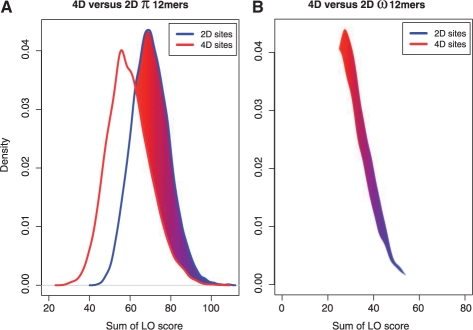
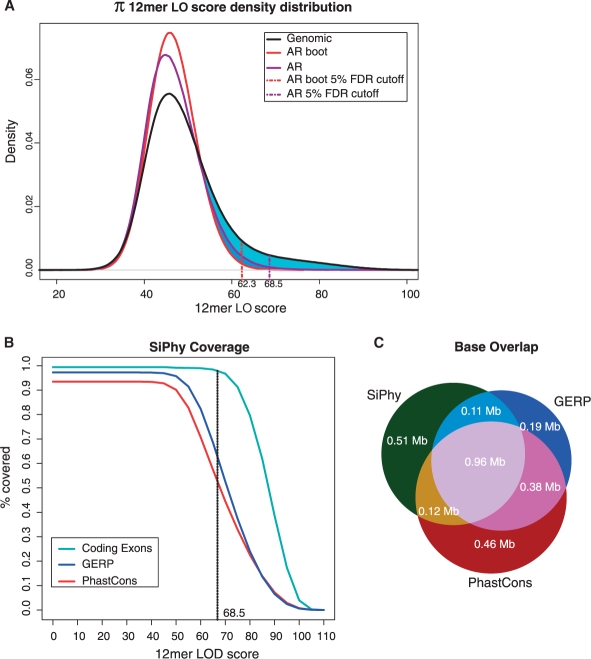
We present a new approach that takes advantage of deeply sequenced clades to identify evolutionary selection by uncovering not only signatures of rate-based conservation but also substitution patterns characteristic of sequence undergoing natural selection. We describe a new statistical method for modeling biased nucleotide substitutions, a learning algorithm for inferring site-specific substitution biases directly from sequence alignments and a hidden Markov model for detecting constrained elements characterized by biased substitutions. We show that the new approach can identify significantly more degenerate constrained sequences than rate-based methods. Applying it to the ENCODE regions, we identify as much as 10.2% of these regions are under selection.
The algorithms are implemented in a Java software package, called SiPhy, freely available at http://www.broadinstitute.org/science/software/.
Supplementary data are available at Bioinformatics online.
比较亲缘关系密切的物种的基因组为识别参考基因组中的功能元件提供了一个强大的工具。已经开发了许多方法来识别跨物种的保守序列;然而,现有方法仅将保守性建模为突变率的降低,而忽略了作用于突变模式的选择。
我们提出了一种新方法,该方法利用深度测序的进化枝来识别进化选择,不仅揭示基于速率的保守性特征,还揭示经历自然选择的序列的替代模式特征。我们描述了一种用于对有偏核苷酸替代进行建模的新统计方法、一种用于直接从序列比对中推断位点特异性替代偏差的学习算法以及一种用于检测以有偏替代为特征的受限元件的隐马尔可夫模型。我们表明,新方法能够比基于速率的方法识别出更多的简并受限序列。将其应用于ENCODE区域,我们发现这些区域中多达10.2%处于选择之下。
这些算法在一个名为SiPhy的Java软件包中实现,可从http://www.broadinstitute.org/science/software/免费获取。
补充数据可在《生物信息学》在线获取。