Rivas Elena, Eddy Sean R
Janelia Farm Research Campus, Howard Hughes Medical Institute, Ashburn, Virginia, United States of America.
PLoS Comput Biol. 2008 Sep 19;4(9):e1000172. doi: 10.1371/journal.pcbi.1000172.
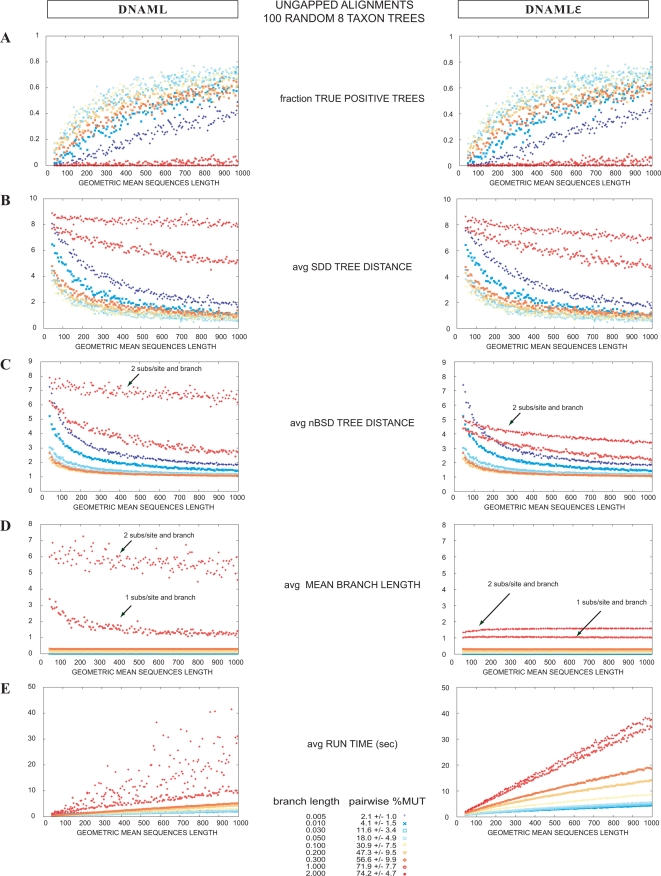
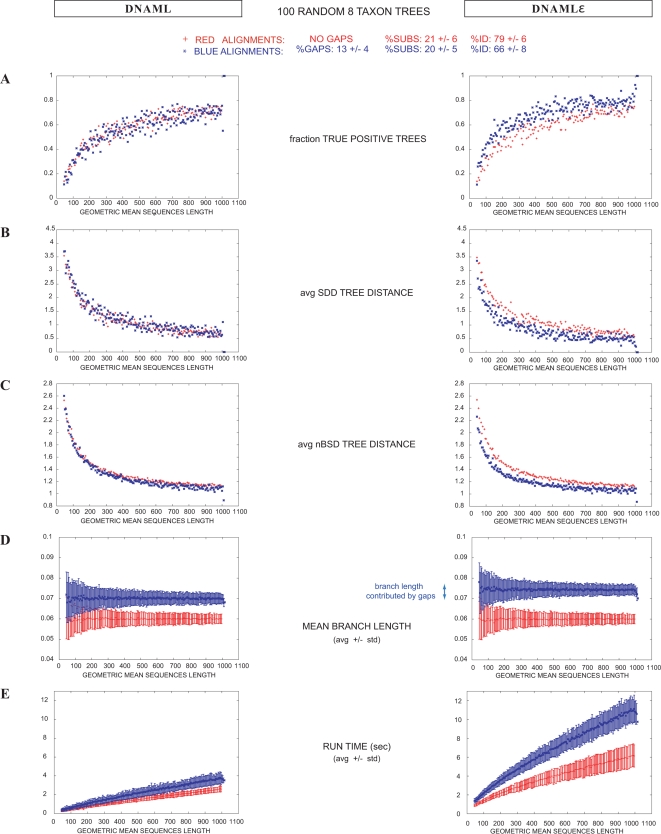
A fundamental task in sequence analysis is to calculate the probability of a multiple alignment given a phylogenetic tree relating the sequences and an evolutionary model describing how sequences change over time. However, the most widely used phylogenetic models only account for residue substitution events. We describe a probabilistic model of a multiple sequence alignment that accounts for insertion and deletion events in addition to substitutions, given a phylogenetic tree, using a rate matrix augmented by the gap character. Starting from a continuous Markov process, we construct a non-reversible generative (birth-death) evolutionary model for insertions and deletions. The model assumes that insertion and deletion events occur one residue at a time. We apply this model to phylogenetic tree inference by extending the program dnaml in phylip. Using standard benchmarking methods on simulated data and a new "concordance test" benchmark on real ribosomal RNA alignments, we show that the extended program dnamlepsilon improves accuracy relative to the usual approach of ignoring gaps, while retaining the computational efficiency of the Felsenstein peeling algorithm.
序列分析中的一项基本任务是,在给出与序列相关的系统发育树以及描述序列随时间如何变化的进化模型的情况下,计算多重比对的概率。然而,最广泛使用的系统发育模型仅考虑残基替换事件。我们描述了一种多重序列比对的概率模型,该模型在给定系统发育树的情况下,除了替换事件外还考虑插入和缺失事件,使用由空位字符扩充的速率矩阵。从连续马尔可夫过程出发,我们构建了一个用于插入和缺失的不可逆生成(生死)进化模型。该模型假设插入和缺失事件一次发生一个残基。我们通过扩展phylip中的程序dnaml,将此模型应用于系统发育树推断。使用模拟数据上的标准基准测试方法以及真实核糖体RNA比对上的新“一致性测试”基准,我们表明扩展后的程序dnamlepsilon相对于忽略空位的常规方法提高了准确性,同时保留了费尔斯滕森剥离算法的计算效率。