Institut für Informatik, Ludwig-Maximilians-Universität München, Amalienstrasse 17, 80333 München, Germany.
Bioinformatics. 2009 Aug 15;25(16):2140-6. doi: 10.1093/bioinformatics/btp353. Epub 2009 Jun 8.
Recent advances in high-throughput technologies have made it possible to investigate not only individual protein interactions, but also the association of these proteins in complexes. So far the focus has been on the prediction of complexes as sets of proteins from the experimental results. The modular substructure and the physical interactions within the protein complexes have been mostly ignored.
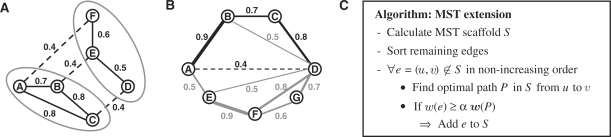
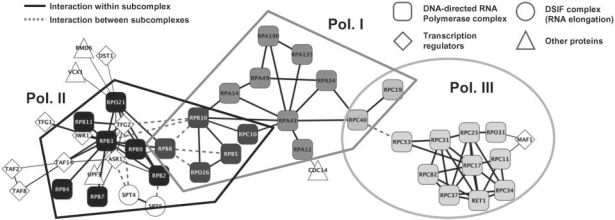
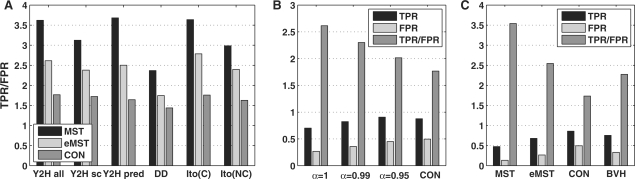
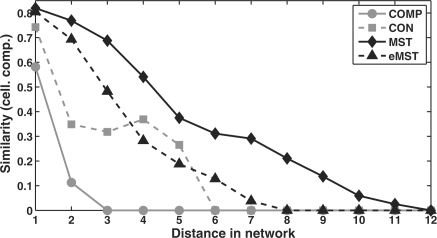
We present an approach for identifying the direct physical interactions and the subcomponent structure of protein complexes predicted from affinity purification assays. Our algorithm calculates the union of all maximum spanning trees from scoring networks for each protein complex to extract relevant interactions. In a subsequent step this network is extended to interactions which are not accounted for by alternative indirect paths. We show that the interactions identified with this approach are more accurate in predicting experimentally derived physical interactions than baseline approaches. Based on these networks, the subcomponent structure of the complexes can be resolved more satisfactorily and subcomplexes can be identified. The usefulness of our method is illustrated on the RNA polymerases for which the modular substructure can be successfully reconstructed.
A Java implementation of the prediction methods and supplementary material are available at http://www.bio.ifi.lmu.de/Complexes/Substructures/.
高通量技术的最新进展不仅使我们能够研究单个蛋白质相互作用,还使我们能够研究这些蛋白质在复合物中的关联。到目前为止,研究的重点一直是从实验结果预测复合物作为蛋白质集合。蛋白质复合物中的模块化子结构和物理相互作用大多被忽略。
我们提出了一种从亲和纯化实验中预测蛋白质复合物的直接物理相互作用和子结构的方法。我们的算法计算了每个蛋白质复合物的评分网络中的所有最大生成树的并集,以提取相关的相互作用。在后续步骤中,将网络扩展到由替代间接路径未考虑的相互作用。我们表明,与基线方法相比,该方法识别的相互作用在预测实验得出的物理相互作用时更准确。基于这些网络,可以更满意地解析复合物的子结构,并识别亚复合物。我们的方法在 RNA 聚合酶上的应用说明了这一点,在 RNA 聚合酶上可以成功重建模块化子结构。
预测方法的 Java 实现和补充材料可在 http://www.bio.ifi.lmu.de/Complexes/Substructures/ 获得。