Tabrizi Sarah J, Langbehn Douglas R, Leavitt Blair R, Roos Raymund Ac, Durr Alexandra, Craufurd David, Kennard Christopher, Hicks Stephen L, Fox Nick C, Scahill Rachael I, Borowsky Beth, Tobin Allan J, Rosas H Diana, Johnson Hans, Reilmann Ralf, Landwehrmeyer Bernhard, Stout Julie C
UCL Institute of Neurology, University College London, Queen Square, London, UK.
Lancet Neurol. 2009 Sep;8(9):791-801. doi: 10.1016/S1474-4422(09)70170-X. Epub 2009 Jul 29.
Huntington's disease (HD) is an autosomal dominant, fully penetrant, neurodegenerative disease that most commonly affects adults in mid-life. Our aim was to identify sensitive and reliable biomarkers in premanifest carriers of mutated HTT and in individuals with early HD that could provide essential methodology for the assessment of therapeutic interventions.
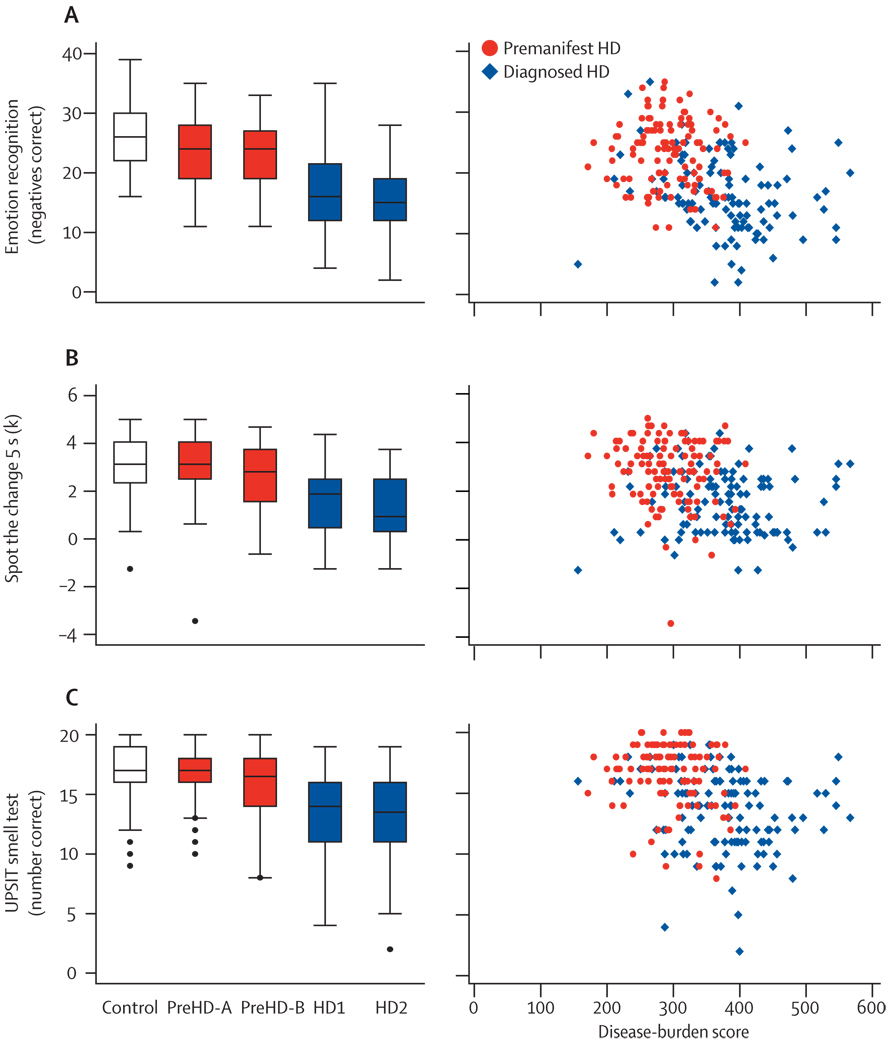
This multicentre study uses an extensive battery of novel assessments, including multi-site 3T MRI, clinical, cognitive, quantitative motor, oculomotor, and neuropsychiatric measures. Blinded analyses were done on the baseline cross-sectional data from 366 individuals: 123 controls, 120 premanifest (pre-HD) individuals, and 123 patients with early HD.
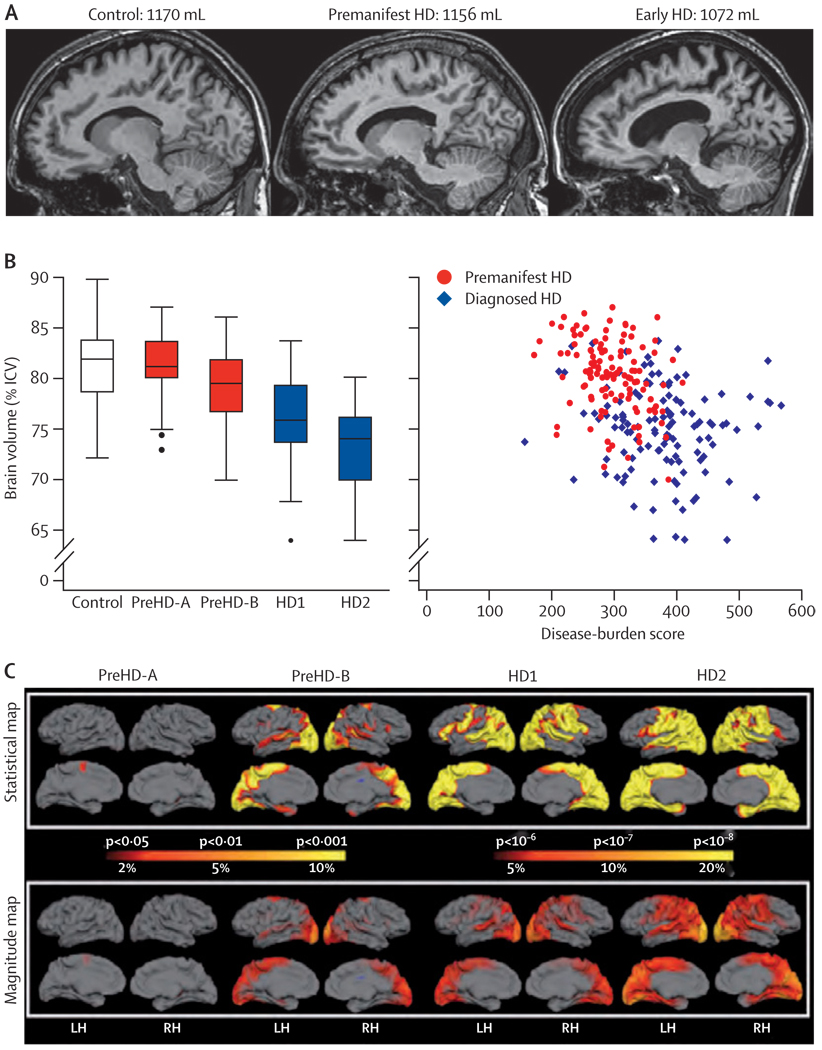
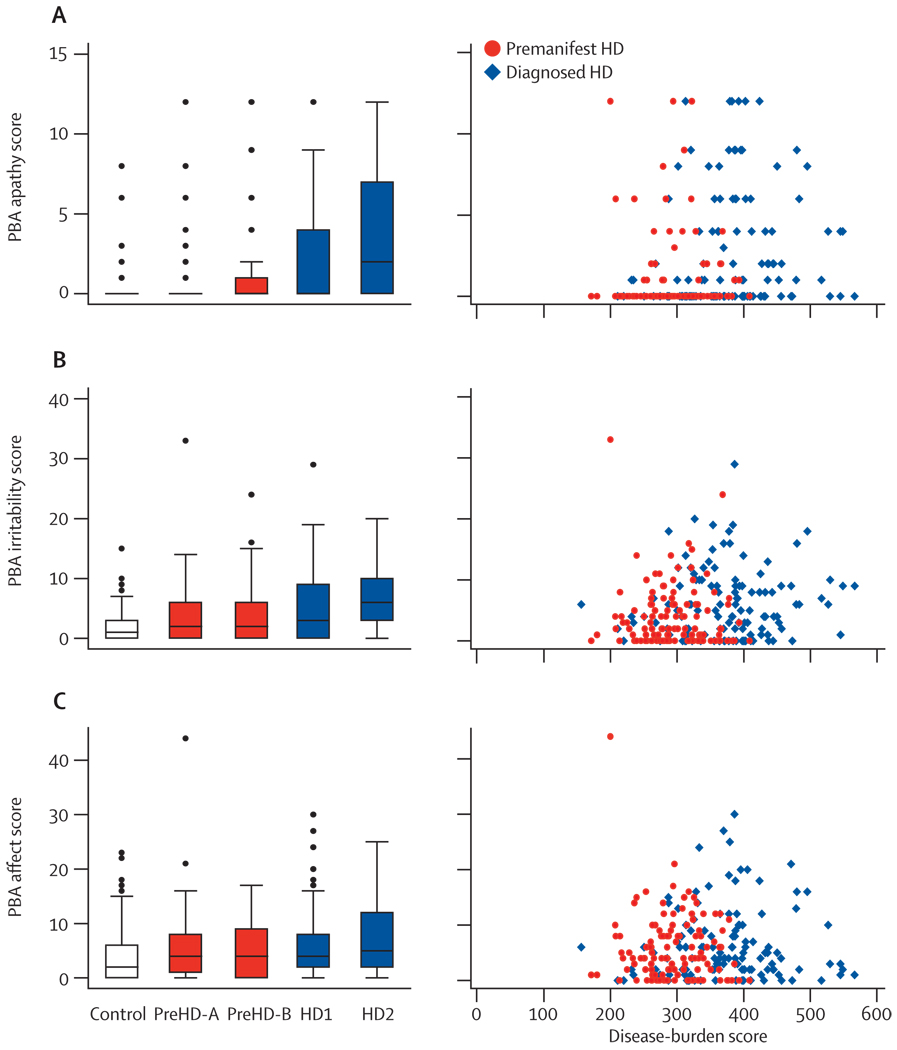
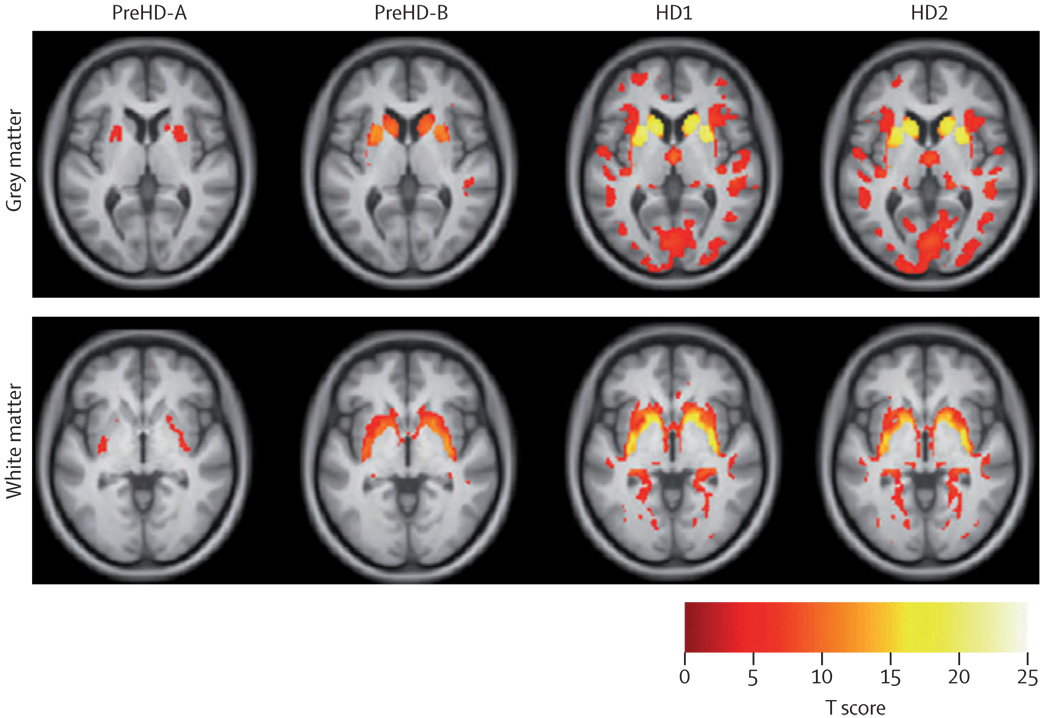
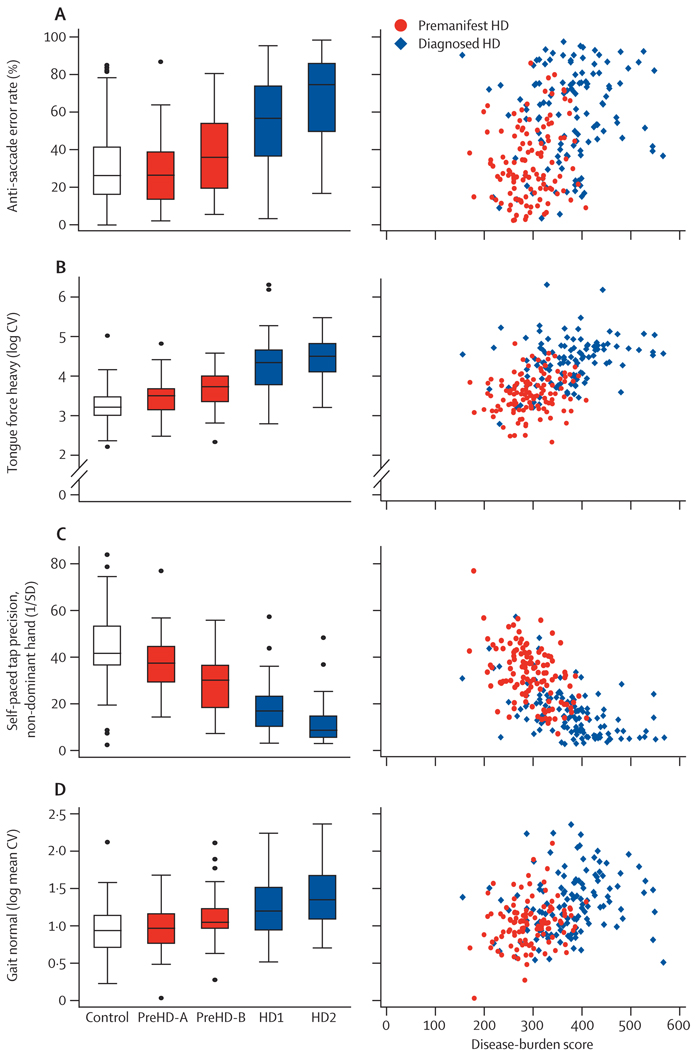
The first participant was enrolled in January, 2008, and all assessments were completed by August, 2008. Cross-sectional analyses identified significant changes in whole-brain volume, regional grey and white matter differences, impairment in a range of voluntary neurophysiological motor, and oculomotor tasks, and cognitive and neuropsychiatric dysfunction in premanifest HD gene carriers with normal motor scores through to early clinical stage 2 disease.
We show the feasibility of rapid data acquisition and the use of multi-site 3T MRI and neurophysiological motor measures in a large multicentre study. Our results provide evidence for quantifiable biological and clinical alterations in HTT expansion carriers compared with age-matched controls. Many parameters differ from age-matched controls in a graded fashion and show changes of increasing magnitude across our cohort, who range from about 16 years from predicted disease diagnosis to early HD. These findings might help to define novel quantifiable endpoints and methods for rapid and reliable data acquisition, which could aid the design of therapeutic trials.
亨廷顿舞蹈症(HD)是一种常染色体显性、完全显性的神经退行性疾病,最常见于中年成年人。我们的目的是在突变型HTT的症状前携带者和早期HD患者中识别敏感且可靠的生物标志物,这些生物标志物可为治疗干预评估提供必要方法。
这项多中心研究采用了一系列广泛的新型评估方法,包括多站点3T磁共振成像(MRI)、临床、认知、定量运动、眼动和神经精神测量。对366名个体的基线横断面数据进行了盲法分析:123名对照者、120名症状前(HD前期)个体和123名早期HD患者。
第一名参与者于2008年1月入组,所有评估于2008年8月完成。横断面分析发现,从运动评分正常的HD基因症状前携带者到临床2期早期疾病患者,全脑体积、区域灰质和白质差异、一系列自主神经生理运动和眼动任务受损以及认知和神经精神功能障碍均有显著变化。
我们展示了在一项大型多中心研究中快速获取数据以及使用多站点3T MRI和神经生理运动测量的可行性。我们的结果为HTT扩展携带者与年龄匹配的对照者相比可量化的生物学和临床改变提供了证据。许多参数与年龄匹配的对照者相比呈分级差异,并在我们的队列中显示出幅度不断增加的变化,我们的队列从预测疾病诊断到早期HD约有16年的跨度。这些发现可能有助于定义新的可量化终点和快速可靠数据采集方法,这有助于治疗试验的设计。