Avila-Rios Santiago, Ormsby Christopher E, Carlson Jonathan M, Valenzuela-Ponce Humberto, Blanco-Heredia Juan, Garrido-Rodriguez Daniela, Garcia-Morales Claudia, Heckerman David, Brumme Zabrina L, Mallal Simon, John Mina, Espinosa Enrique, Reyes-Teran Gustavo
Center for Research in Infectious Diseases, National Institute of Respiratory Diseases, Mexico City, Mexico.
Retrovirology. 2009 Aug 10;6:72. doi: 10.1186/1742-4690-6-72.
Mounting evidence indicates that HLA-mediated HIV evolution follows highly stereotypic pathways that result in HLA-associated footprints in HIV at the population level. However, it is not known whether characteristic HLA frequency distributions in different populations have resulted in additional unique footprints.
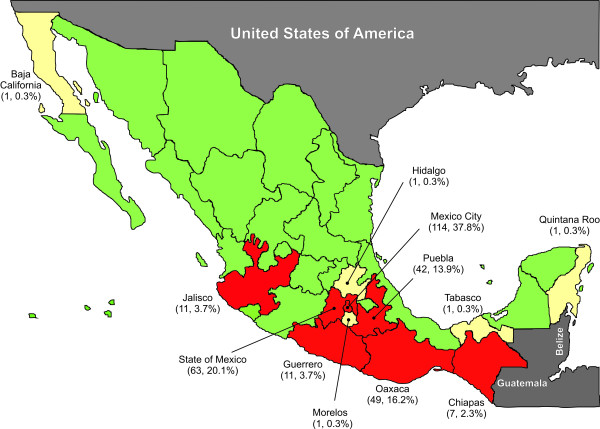
The phylogenetic dependency network model was applied to assess HLA-mediated evolution in datasets of HIV pol sequences from free plasma viruses and peripheral blood mononuclear cell (PBMC)-integrated proviruses in an immunogenetically unique cohort of Mexican individuals. Our data were compared with data from the IHAC cohort, a large multi-center cohort of individuals from Canada, Australia and the USA.
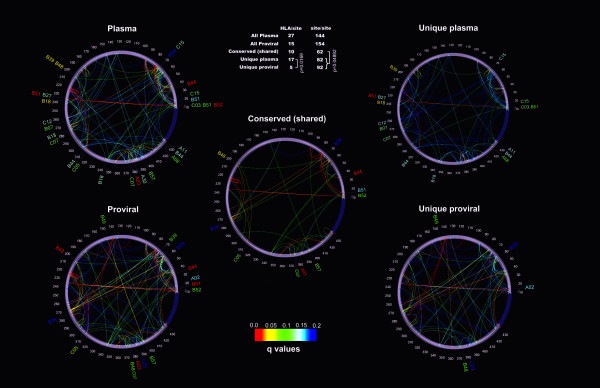
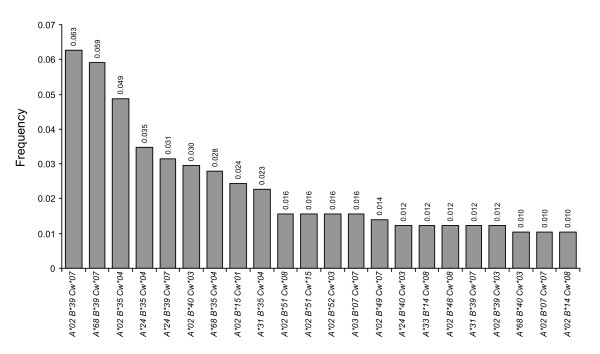
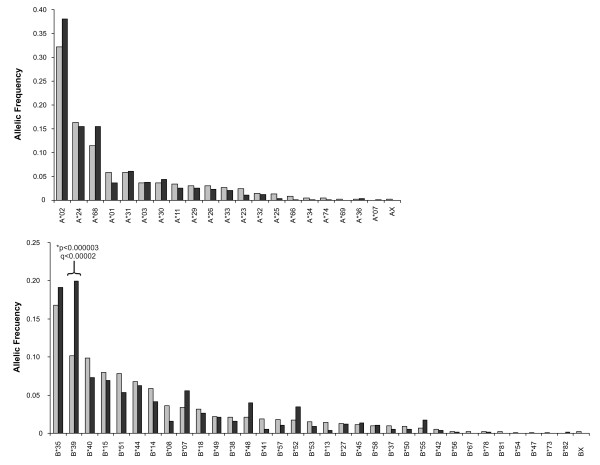
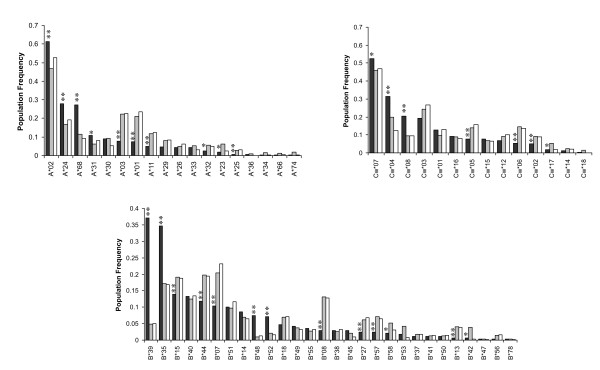
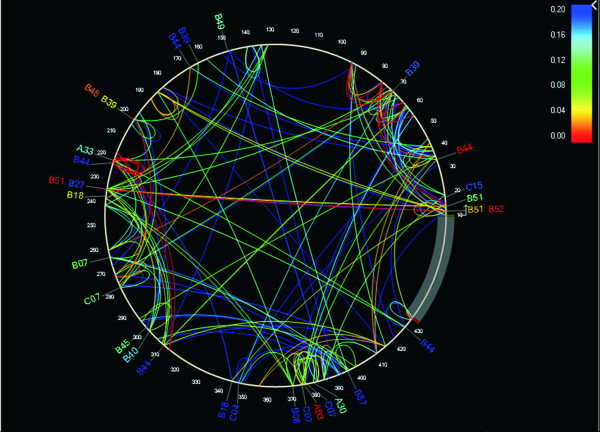
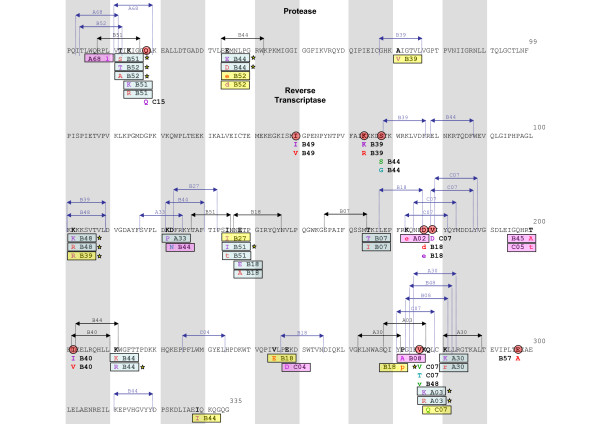
Forty three different HLA-HIV codon associations representing 30 HLA-HIV codon pairs were observed in the Mexican cohort (q < 0.2). Strikingly, 23 (53%) of these associations differed from those observed in the well-powered IHAC cohort, strongly suggesting the existence of unique characteristics in HLA-mediated HIV evolution in the Mexican cohort. Furthermore, 17 of the 23 novel associations involved HLA alleles whose frequencies were not significantly different from those in IHAC, suggesting that their detection was not due to increased statistical power but to differences in patterns of epitope targeting. Interestingly, the consensus differed in four positions between the two cohorts and three of these positions could be explained by HLA-associated selection. Additionally, different HLA-HIV codon associations were seen when comparing HLA-mediated selection in plasma viruses and PBMC archived proviruses at the population level, with a significantly lower number of associations in the proviral dataset.
Our data support universal HLA-mediated HIV evolution at the population level, resulting in detectable HLA-associated footprints in the circulating virus. However, it also strongly suggests that unique genetic backgrounds in different HIV-infected populations may influence HIV evolution in a particular direction as particular HLA-HIV codon associations are determined by specific HLA frequency distributions. Our analysis also suggests a dynamic HLA-associated evolution in HIV with fewer HLA-HIV codon associations observed in the proviral compartment, which is likely enriched in early archived HIV sequences, compared to the plasma virus compartment. These results highlight the importance of comparative HIV evolutionary studies in immunologically different populations worldwide.
越来越多的证据表明,HLA介导的HIV进化遵循高度刻板的途径,在群体水平上导致HIV中出现与HLA相关的印记。然而,尚不清楚不同人群中特征性的HLA频率分布是否导致了额外的独特印记。
应用系统发育依赖网络模型,评估墨西哥个体免疫遗传独特队列中游离血浆病毒和外周血单核细胞(PBMC)整合前病毒的HIV pol序列数据集中HLA介导的进化。将我们的数据与IHAC队列的数据进行比较,IHAC队列是一个来自加拿大、澳大利亚和美国的大型多中心个体队列。
在墨西哥队列中观察到43种不同的HLA-HIV密码子关联,代表30个HLA-HIV密码子对(q < 0.2)。令人惊讶的是,其中23种(53%)关联与在样本量充足的IHAC队列中观察到的不同,强烈表明墨西哥队列中HLA介导的HIV进化存在独特特征。此外,23种新关联中的17种涉及HLA等位基因,其频率与IHAC队列中的频率无显著差异,这表明它们的检测不是由于统计效力增加,而是由于表位靶向模式的差异。有趣的是,两个队列之间在四个位置上的共识不同,其中三个位置可以用HLA相关选择来解释。此外,在群体水平上比较血浆病毒和PBMC存档前病毒中HLA介导的选择时,观察到不同的HLA-HIV密码子关联,前病毒数据集中的关联数量明显较少。
我们的数据支持群体水平上普遍存在的HLA介导的HIV进化,导致循环病毒中出现可检测的与HLA相关的印记。然而,这也强烈表明,不同HIV感染人群中的独特遗传背景可能会影响HIV朝着特定方向进化,因为特定的HLA-HIV密码子关联由特定的HLA频率分布决定。我们的分析还表明,HIV中存在动态的HLA相关进化,与血浆病毒区室相比,在前病毒区室中观察到的HLA-HIV密码子关联较少,前病毒区室可能富集了早期存档的HIV序列。这些结果突出了全球免疫不同人群中比较HIV进化研究的重要性。