Kubicki James D, Halada Gary P, Jha Prashant, Phillips Brian L
Department of Geosciences, The Pennsylvania State University, University Park, PA 16802, USA.
Chem Cent J. 2009 Aug 18;3:10. doi: 10.1186/1752-153X-3-10.
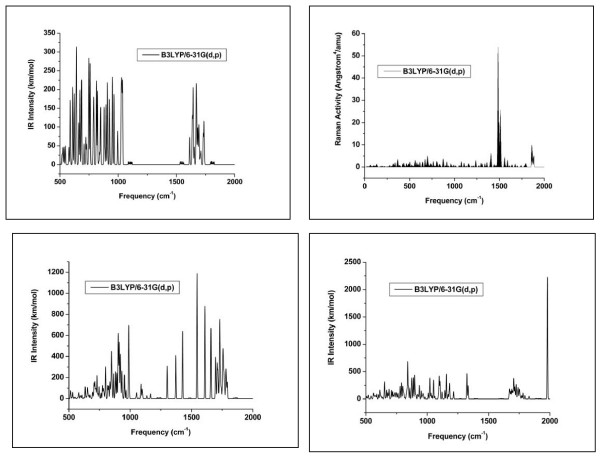
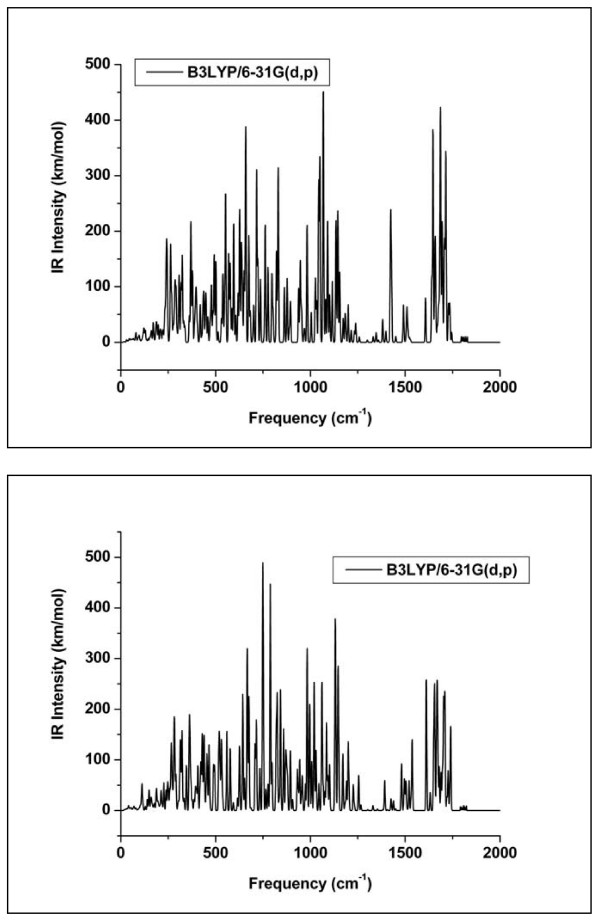
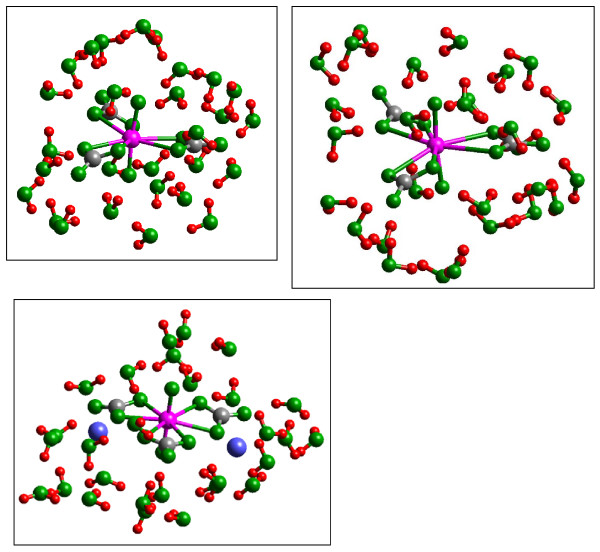
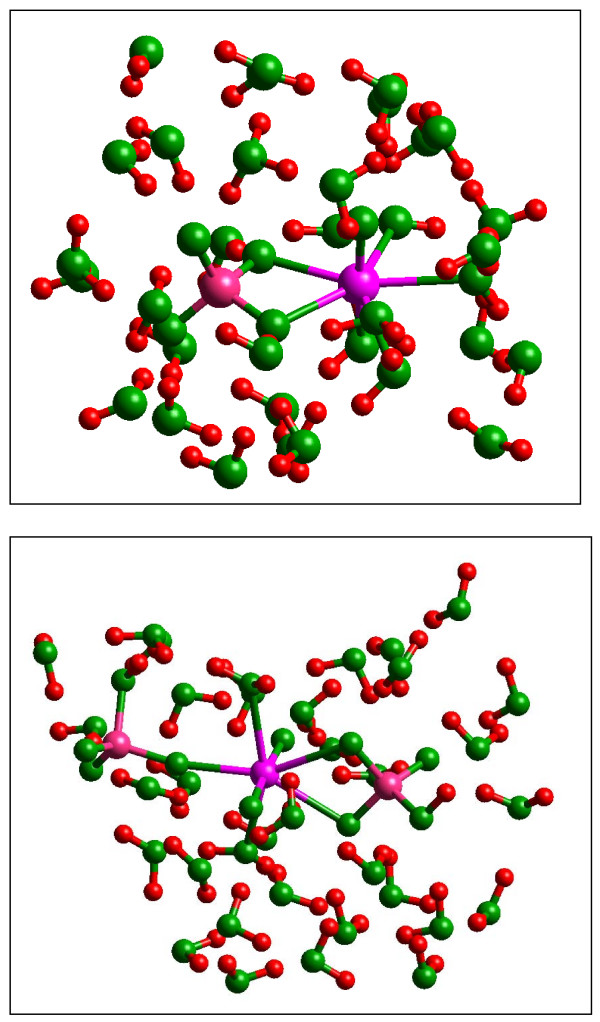
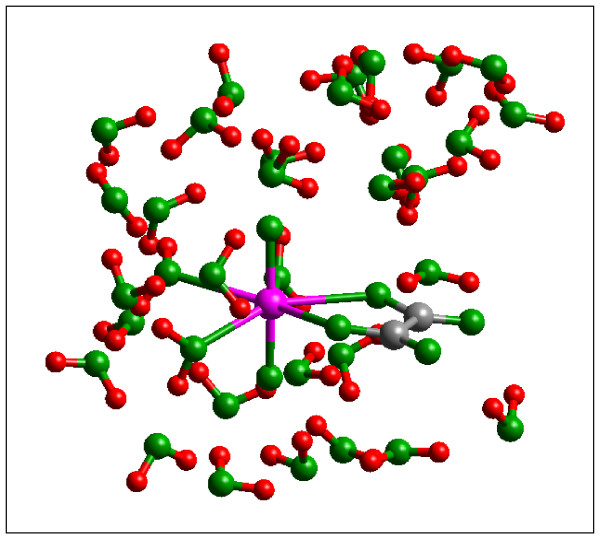
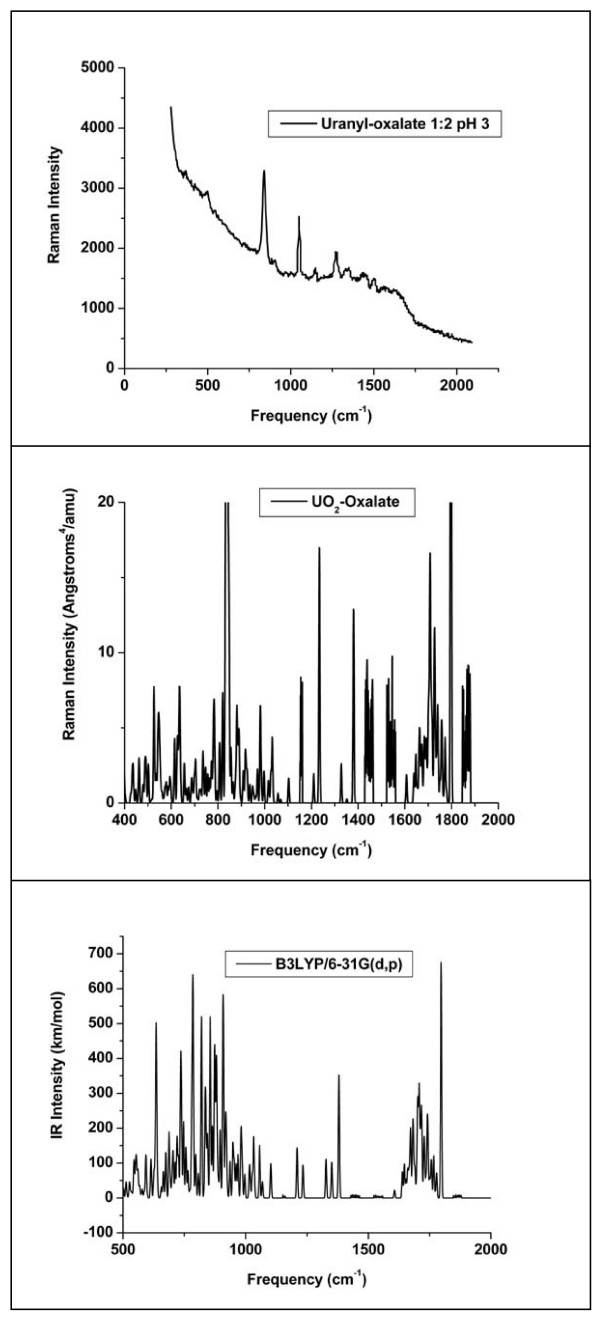
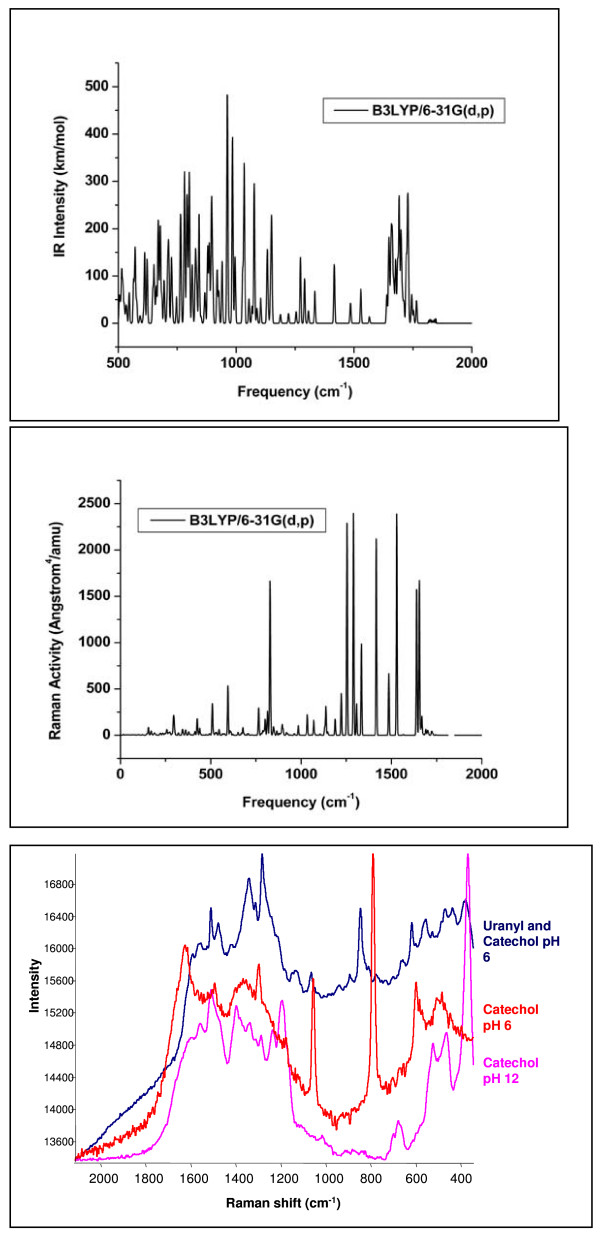
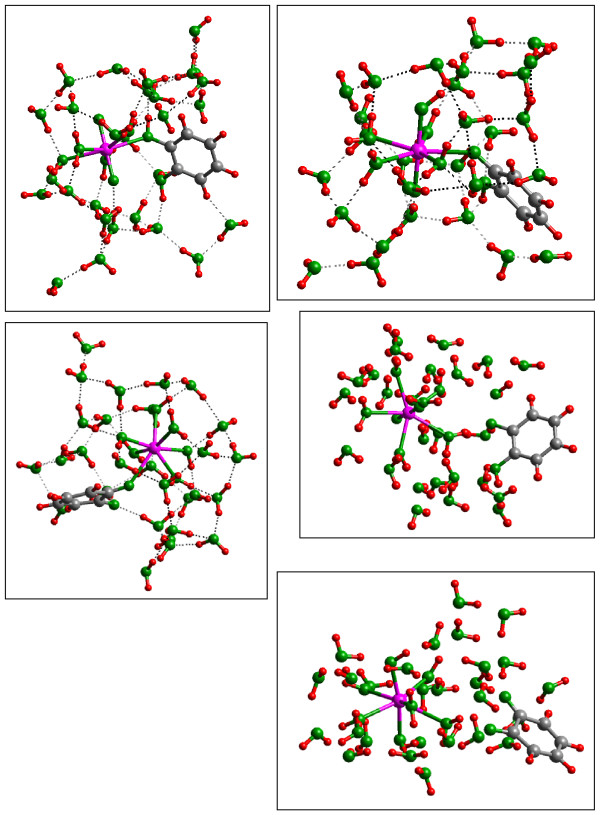
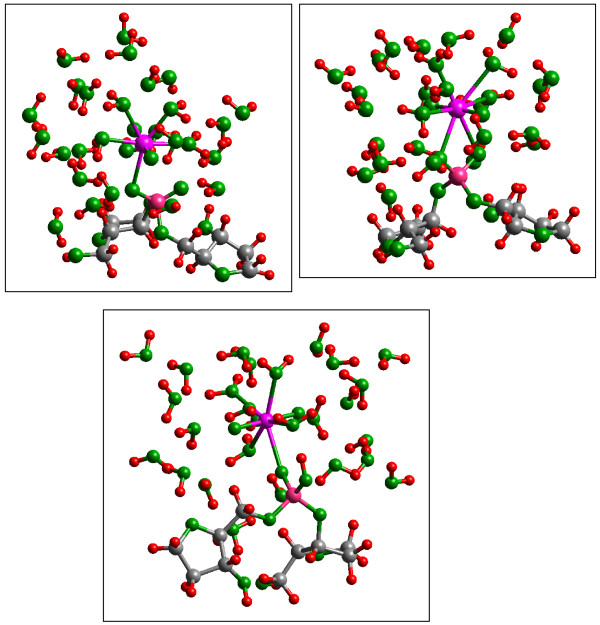
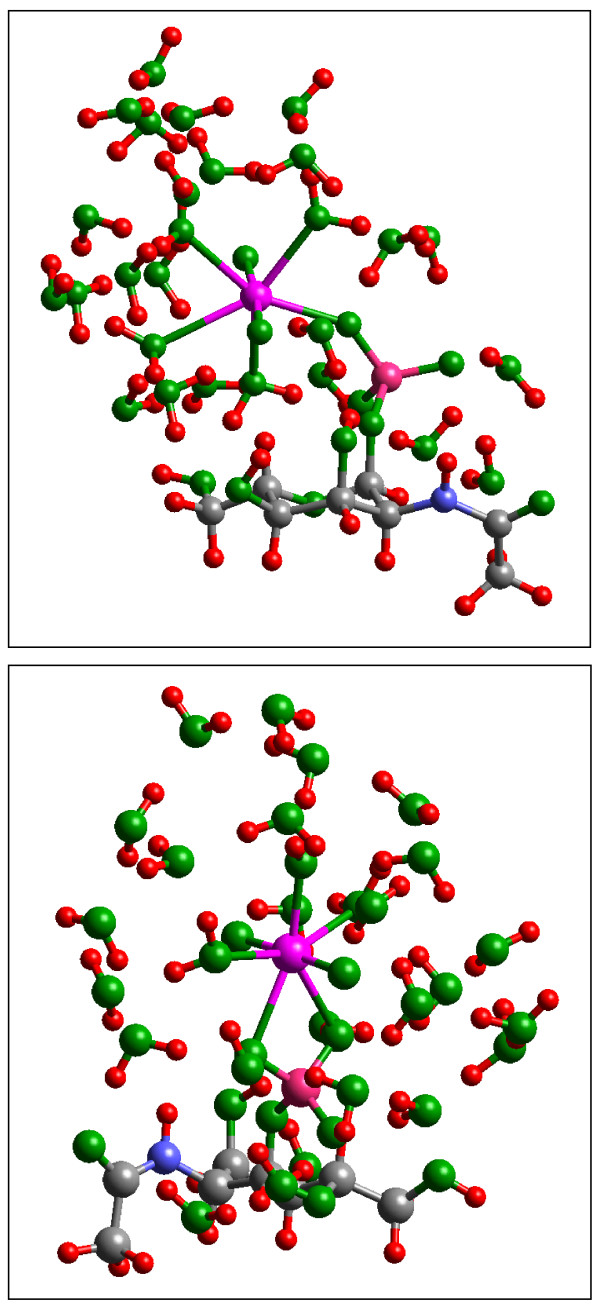
Quantum mechanical calculations were performed on a variety of uranium species representing U(VI), U(V), U(IV), U-carbonates, U-phosphates, U-oxalates, U-catecholates, U-phosphodiesters, U-phosphorylated N-acetyl-glucosamine (NAG), and U-2-Keto-3-doxyoctanoate (KDO) with explicit solvation by H2O molecules. These models represent major U species in natural waters and complexes on bacterial surfaces. The model results are compared to observed EXAFS, IR, Raman and NMR spectra.
Agreement between experiment and theory is acceptable in most cases, and the reasons for discrepancies are discussed. Calculated Gibbs free energies are used to constrain which configurations are most likely to be stable under circumneutral pH conditions. Reduction of U(VI) to U(IV) is examined for the U-carbonate and U-catechol complexes.
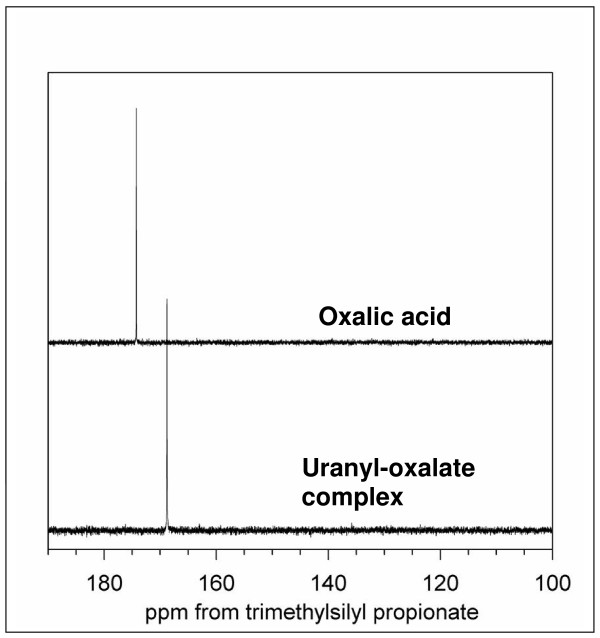
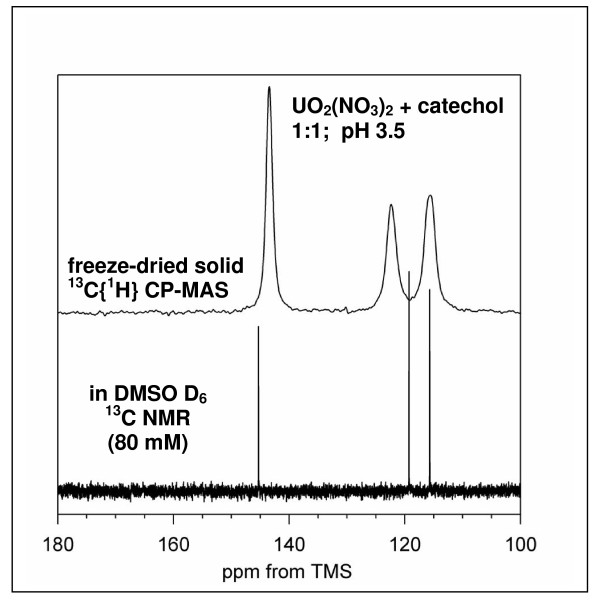
Results on the potential energy differences between U(V)- and U(IV)-carbonate complexes suggest that the cause of slower disproportionation in this system is electrostatic repulsion between UO2 [CO3]3(5-) ions that must approach one another to form U(VI) and U(IV) rather than a change in thermodynamic stability. Calculations on U-catechol species are consistent with the observation that UO2(2+) can oxidize catechol and form quinone-like species. In addition, outer-sphere complexation is predicted to be the most stable for U-catechol interactions based on calculated energies and comparison to 13C NMR spectra. Outer-sphere complexes (i.e., ion pairs bridged by water molecules) are predicted to be comparable in Gibbs free energy to inner-sphere complexes for a model carboxylic acid. Complexation of uranyl to phosphorus-containing groups in extracellular polymeric substances is predicted to favor phosphonate groups, such as that found in phosphorylated NAG, rather than phosphodiesters, such as those in nucleic acids.
对多种铀物种进行了量子力学计算,这些物种代表U(VI)、U(V)、U(IV)、铀碳酸盐、铀磷酸盐、铀草酸盐、铀儿茶酚盐、铀磷酸二酯、铀磷酸化N - 乙酰葡糖胺(NAG)和铀2 - 酮 - 3 - 脱氧辛酸酯(KDO),并通过水分子进行明确的溶剂化处理。这些模型代表了天然水体中的主要铀物种以及细菌表面的配合物。将模型结果与观测到的扩展X射线吸收精细结构(EXAFS)、红外(IR)、拉曼和核磁共振(NMR)光谱进行了比较。
在大多数情况下,实验与理论之间的一致性是可以接受的,并对差异的原因进行了讨论。计算得到的吉布斯自由能用于确定在接近中性pH条件下哪些构型最有可能是稳定的。研究了铀碳酸盐和铀儿茶酚配合物中U(VI)还原为U(IV)的情况。
U(V) - 和U(IV) - 碳酸盐配合物之间势能差的结果表明,该系统中歧化反应较慢的原因是UO2 [CO3]3(5-)离子之间的静电排斥,这些离子必须相互靠近才能形成U(VI)和U(IV),而不是热力学稳定性的变化。对铀儿茶酚物种的计算与UO2(2+)可以氧化儿茶酚并形成醌类物种的观测结果一致。此外,基于计算能量并与13C NMR光谱比较,预测外层配合作用对于铀儿茶酚相互作用是最稳定的。对于一种模型羧酸,预测外层配合物(即由水分子桥接的离子对)的吉布斯自由能与内层配合物相当。预计铀酰与细胞外聚合物中含磷基团的络合作用有利于膦酸酯基团,如在磷酸化NAG中发现的基团,而不是磷酸二酯,如核酸中的那些基团。