Zeviani M, Bresolin N, Gellera C, Bordoni A, Pannacci M, Amati P, Moggio M, Servidei S, Scarlato G, DiDonato S
National Neurological Institute, Carlo Besta, Department of Biochemistry and Genetics, State University School of Medicine, Milan, Italy.
Am J Hum Genet. 1990 Dec;47(6):904-14.
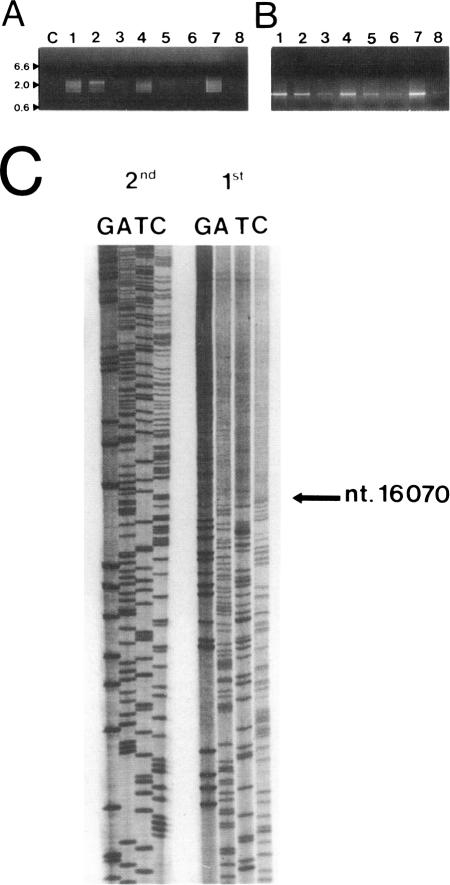
We studied several affected and one nonaffected individuals belonging to three unrelated pedigrees. The pathological trait was an autosomal dominant mitochondrial myopathy due to large-scale multiple deletions of the mitochondrial genome. Clinically, symptomatic patients had progressive external ophthalmoplegia, muscle weakness and wasting, sensorineural hypoacusia, and, in some cases, vestibular areflexia and tremor. The muscle biopsies of all patients examined showed ragged-red fibers, neurogenic changes, and a partially decreased histochemical reaction to cytochrome c oxidase. Multiple mtDNA heteroplasmy was detected in the patients by both Southern blot analysis and PCR amplification, whereas the unaffected individual had the normal homoplasmic hybridization pattern. These findings confirm and add further details to the existence of a new human disease--defined clinically as a mitochondrial myopathy, genetically as a Mendelian autosomal dominant trait, and molecularly by the accumulation of multiple, large-scale deletions of the mitochondrial genome--that is due to impaired nuclear control during mtDNA replication.
我们研究了来自三个无亲缘关系家系的几名患病个体和一名未患病个体。病理特征为常染色体显性线粒体肌病,由线粒体基因组的大规模多重缺失引起。临床上,有症状的患者表现为进行性眼外肌麻痹、肌肉无力和萎缩、感觉神经性听力减退,部分病例还伴有前庭反射消失和震颤。所有接受检查的患者的肌肉活检均显示有破碎红纤维、神经源性改变以及对细胞色素c氧化酶的组织化学反应部分减弱。通过Southern印迹分析和PCR扩增在患者中检测到多个线粒体DNA异质性,而未患病个体具有正常的同质性杂交模式。这些发现证实并进一步详细说明了一种新的人类疾病的存在——临床上定义为线粒体肌病,遗传学上为孟德尔常染色体显性性状,分子水平上则是由于线粒体基因组的多个大规模缺失的积累——这是由于线粒体DNA复制过程中核控制受损所致。