Johnson Philip L F, Slatkin Montgomery
Biophysics Graduate Group, University of California Berkeley, Berkeley, California, United States of America.
PLoS Genet. 2009 Oct;5(10):e1000674. doi: 10.1371/journal.pgen.1000674. Epub 2009 Oct 2.
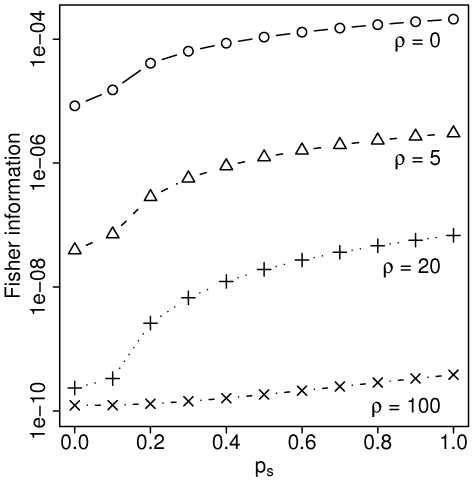
Metagenomic sequencing projects from environments dominated by a small number of species produce genome-wide population samples. We present a two-site composite likelihood estimator of the scaled recombination rate, rho = 2N(e)c, that operates on metagenomic assemblies in which each sequenced fragment derives from a different individual. This new estimator properly accounts for sequencing error, as quantified by per-base quality scores, and missing data, as inferred from the placement of reads in a metagenomic assembly. We apply our estimator to data from a sludge metagenome project to demonstrate how this method will elucidate the rates of exchange of genetic material in natural microbial populations. Surprisingly, for a fixed amount of sequencing, this estimator has lower variance than similar methods that operate on more traditional population genetic samples of comparable size. In addition, we can infer variation in recombination rate across the genome because metagenomic projects sample genetic diversity genome-wide, not just at particular loci. The method itself makes no assumption specific to microbial populations, opening the door for application to any mixed population sample where the number of individuals sampled is much greater than the number of fragments sequenced.
来自少数物种占主导的环境的宏基因组测序项目会产生全基因组的群体样本。我们提出了一种关于缩放重组率rho = 2N(e)c的双位点复合似然估计器,该估计器作用于宏基因组组装体,其中每个测序片段都来自不同的个体。这个新的估计器能够恰当地考虑测序错误(由每个碱基的质量分数量化)以及缺失数据(从宏基因组组装体中读取片段的位置推断)。我们将我们的估计器应用于污泥宏基因组项目的数据,以展示这种方法将如何阐明自然微生物群体中遗传物质的交换率。令人惊讶的是,对于固定的测序量,该估计器的方差比在类似大小的更传统群体遗传样本上运行的类似方法更低。此外,由于宏基因组项目在全基因组范围内对遗传多样性进行采样,而不仅仅是在特定位点,我们可以推断出全基因组范围内重组率的变化。该方法本身并不对微生物群体做特定假设,为应用于任何个体采样数量远大于测序片段数量的混合群体样本打开了大门。