Berger Swetlana, Schlather Martin, de los Campos Gustavo, Weigend Steffen, Preisinger Rudolf, Erbe Malena, Simianer Henner
Animal Breeding and Genetics Group, Department of Animal Sciences, Georg-August-University, Goettingen, Germany.
School of Business Informatics and Mathematics, University of Mannheim, Mannheim, Germany.
PLoS One. 2015 Oct 30;10(10):e0141216. doi: 10.1371/journal.pone.0141216. eCollection 2015.
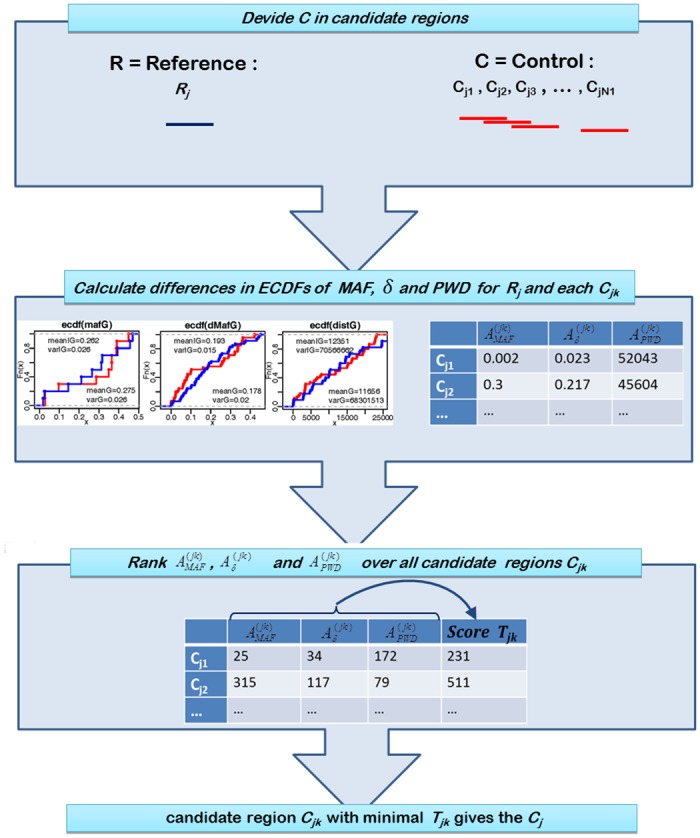
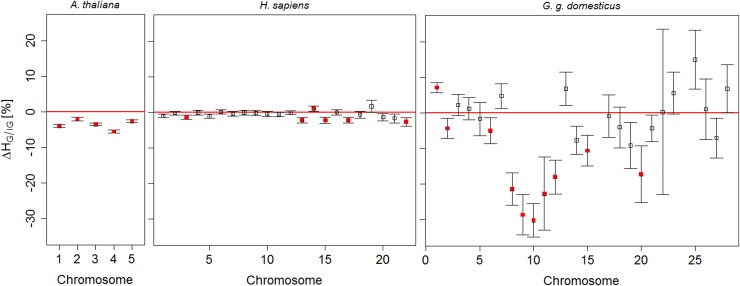
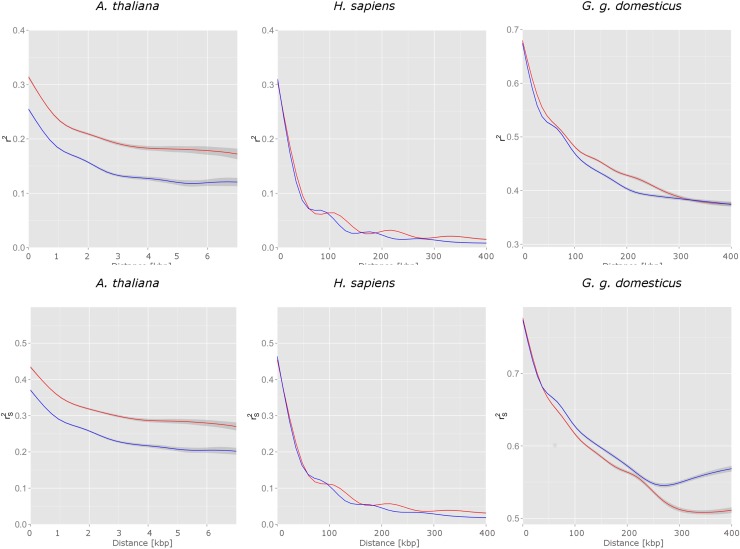
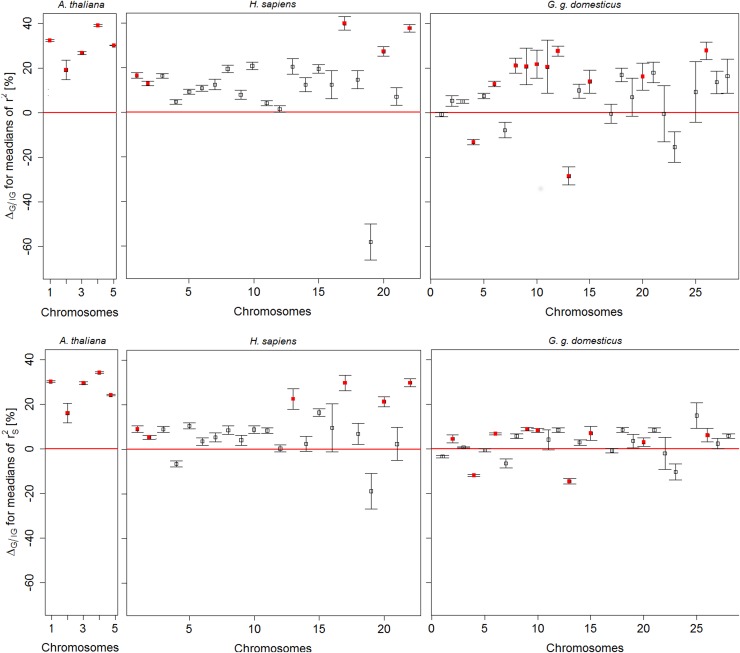
The understanding of non-random association between loci, termed linkage disequilibrium (LD), plays a central role in genomic research. Since causal mutations are generally not included in genomic marker data, LD between those and available markers is essential for capturing the effects of causal loci on localizing genes responsible for traits. Thus, the interpretation of association studies requires a detailed knowledge of LD patterns. It is well known that most LD measures depend on minor allele frequencies (MAF) of the considered loci and the magnitude of LD is influenced by the physical distances between loci. In the present study, a procedure to compare the LD structure between genomic regions comprising several markers each is suggested. The approach accounts for different scaling factors, namely the distribution of MAF, the distribution of pair-wise differences in MAF, and the physical extent of compared regions, reflected by the distribution of pair-wise physical distances. In the first step, genomic regions are matched based on similarity in these scaling factors. In the second step, chromosome- and genome-wide significance tests for differences in medians of LD measures in each pair are performed. The proposed framework was applied to test the hypothesis that the average LD is different in genic and non-genic regions. This was tested with a genome-wide approach with data sets for humans (Homo sapiens), a highly selected chicken line (Gallus gallus domesticus) and the model plant Arabidopsis thaliana. In all three data sets we found a significantly higher level of LD in genic regions compared to non-genic regions. About 31% more LD was detected genome-wide in genic compared to non-genic regions in Arabidopsis thaliana, followed by 13.6% in human and 6% chicken. Chromosome-wide comparison discovered significant differences on all 5 chromosomes in Arabidopsis thaliana and on one third of the human and of the chicken chromosomes.
对基因座之间非随机关联的理解,即连锁不平衡(LD),在基因组研究中起着核心作用。由于因果突变通常不包含在基因组标记数据中,这些因果突变与可用标记之间的连锁不平衡对于捕捉因果基因座对定位性状相关基因的影响至关重要。因此,关联研究的解释需要对连锁不平衡模式有详细的了解。众所周知,大多数连锁不平衡度量取决于所考虑基因座的次要等位基因频率(MAF),并且连锁不平衡的程度受基因座之间物理距离的影响。在本研究中,提出了一种比较每个包含多个标记的基因组区域之间连锁不平衡结构的方法。该方法考虑了不同的缩放因子,即次要等位基因频率的分布、次要等位基因频率中成对差异的分布,以及由成对物理距离的分布所反映的比较区域的物理范围。第一步,基于这些缩放因子的相似性对基因组区域进行匹配。第二步,对每对中连锁不平衡度量中位数的差异进行全染色体和全基因组显著性检验。所提出的框架被应用于检验基因区域和非基因区域中平均连锁不平衡不同的假设。这通过全基因组方法,利用人类(智人)、一个高度选育的鸡品系(家鸡)和模式植物拟南芥的数据集进行了检验。在所有三个数据集中,我们发现基因区域中的连锁不平衡水平显著高于非基因区域。在拟南芥中,全基因组范围内检测到基因区域中的连锁不平衡比非基因区域多约31%,其次是人类中的13.6%和鸡中的6%。全染色体比较发现,拟南芥的所有5条染色体以及人类和鸡染色体的三分之一上存在显著差异。