Familial Cancer Clinic, Gastroenterology Dept, Meir Hospital, Kfar Saba, Israel.
Orphanet J Rare Dis. 2009 Oct 12;4:22. doi: 10.1186/1750-1172-4-22.
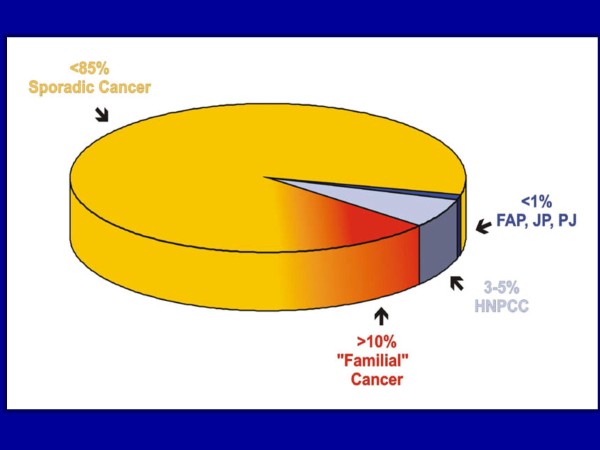
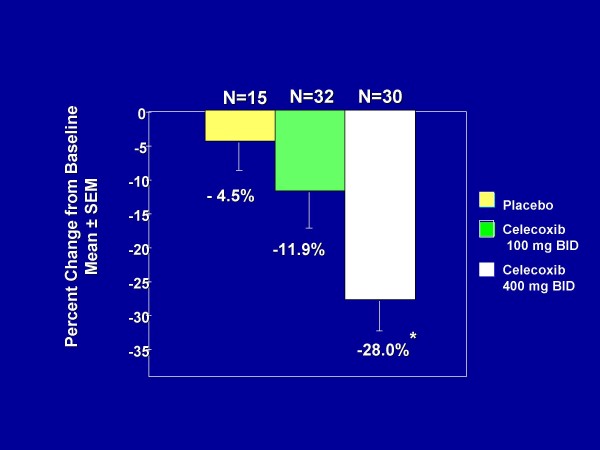
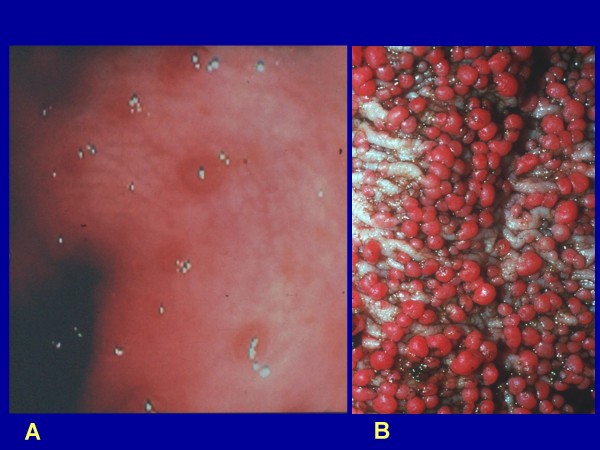
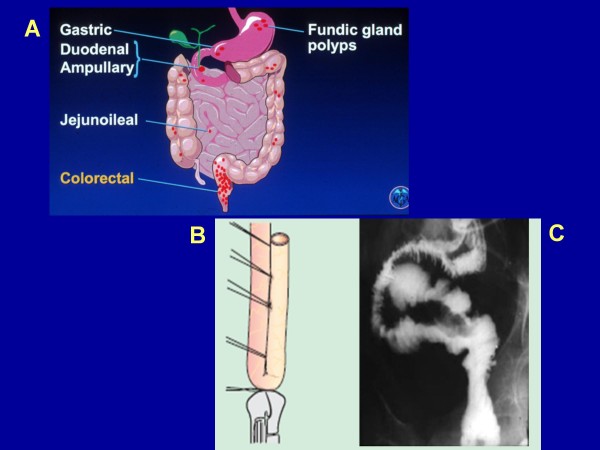
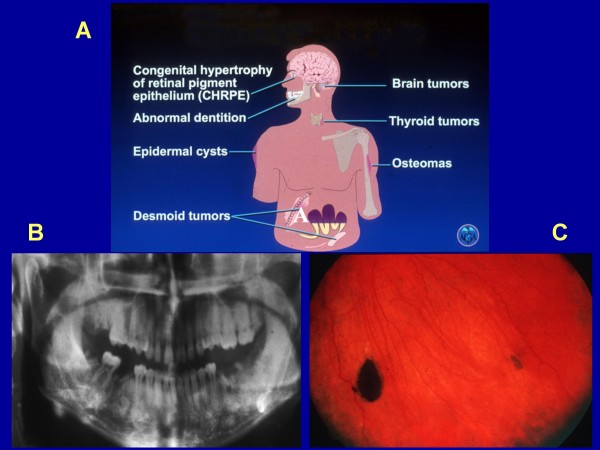
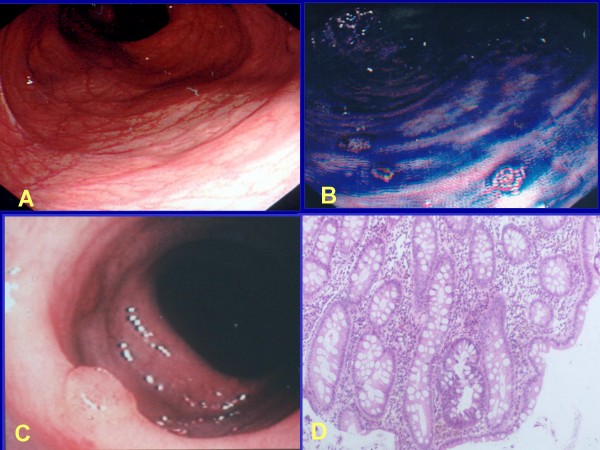
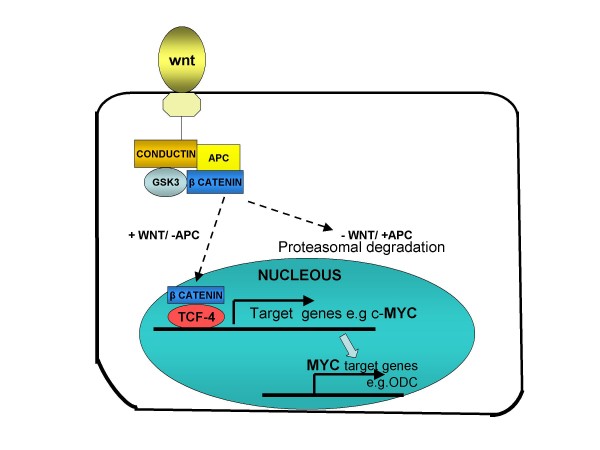
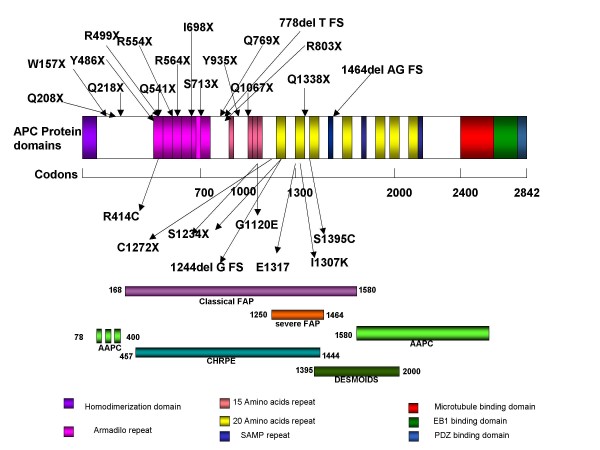
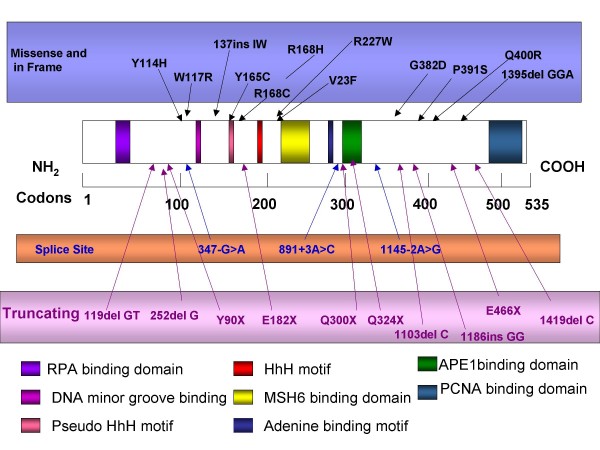
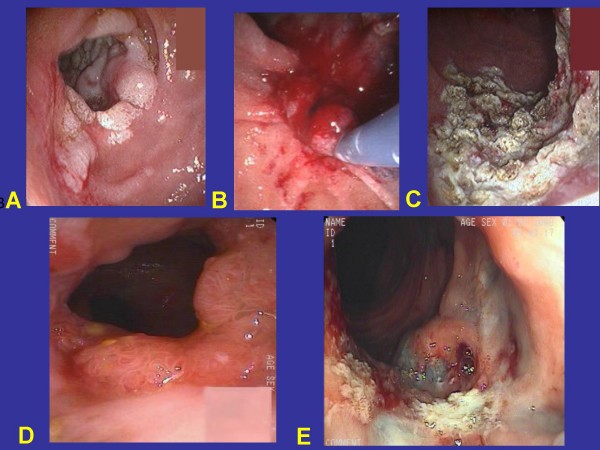
Familial adenomatous polyposis (FAP) is characterized by the development of many tens to thousands of adenomas in the rectum and colon during the second decade of life. FAP has an incidence at birth of about 1/8,300, it manifests equally in both sexes, and accounts for less than 1% of colorectal cancer (CRC) cases. In the European Union, prevalence has been estimated at 1/11,300-37,600. Most patients are asymptomatic for years until the adenomas are large and numerous, and cause rectal bleeding or even anemia, or cancer develops. Generally, cancers start to develop a decade after the appearance of the polyps. Nonspecific symptoms may include constipation or diarrhea, abdominal pain, palpable abdominal masses and weight loss. FAP may present with some extraintestinal manifestations such as osteomas, dental abnormalities (unerupted teeth, congenital absence of one or more teeth, supernumerary teeth, dentigerous cysts and odontomas), congenital hypertrophy of the retinal pigment epithelium (CHRPE), desmoid tumors, and extracolonic cancers (thyroid, liver, bile ducts and central nervous system). A less aggressive variant of FAP, attenuated FAP (AFAP), is characterized by fewer colorectal adenomatous polyps (usually 10 to 100), later age of adenoma appearance and a lower cancer risk. Some lesions (skull and mandible osteomas, dental abnormalities, and fibromas on the scalp, shoulders, arms and back) are indicative of the Gardner variant of FAP. Classic FAP is inherited in an autosomal dominant manner and results from a germline mutation in the adenomatous polyposis (APC) gene. Most patients (~70%) have a family history of colorectal polyps and cancer. In a subset of individuals, a MUTYH mutation causes a recessively inherited polyposis condition, MUTYH-associated polyposis (MAP), which is characterized by a slightly increased risk of developing CRC and polyps/adenomas in both the upper and lower gastrointestinal tract. Diagnosis is based on a suggestive family history, clinical findings, and large bowel endoscopy or full colonoscopy. Whenever possible, the clinical diagnosis should be confirmed by genetic testing. When the APC mutation in the family has been identified, genetic testing of all first-degree relatives should be performed. Presymptomatic and prenatal (amniocentesis and chorionic villous sampling), and even preimplantation genetic testing is possible. Referral to a geneticist or genetic counselor is mandatory. Differential diagnoses include other disorders causing multiple polyps (such as Peutz-Jeghers syndrome, familial juvenile polyps or hyperplastic polyposis, hereditary mixed polyposis syndromes, and Lynch syndrome). Cancer prevention and maintaining a good quality of life are the main goals of management and regular and systematic follow-up and supportive care should be offered to all patients. By the late teens or early twenties, colorectal cancer prophylactic surgery is advocated. The recommended alternatives are total proctocolectomy and ileoanal pouch or ileorectal anastomosis for AFAP. Duodenal cancer and desmoids are the two main causes of mortality after total colectomy, they need to be identified early and treated. Upper endoscopy is necessary for surveillance to reduce the risk of ampullary and duodenal cancer. Patients with progressive tumors and unresectable disease may respond or stabilize with a combination of cytotoxic chemotherapy and surgery (when possible to perform). Adjunctive therapy with celecoxib has been approved by the US Food and Drug Administration and the European Medicines Agency in patients with FAP. Individuals with FAP carry a 100% risk of CRC; however, this risk is reduced significantly when patients enter a screening-treatment program.
家族性腺瘤性息肉病(FAP)的特征是在生命的第二个十年中直肠和结肠中发展出数十到数千个腺瘤。FAP 的出生时发病率约为 1/8300,男女发病率均等,占结直肠癌(CRC)病例的不到 1%。在欧盟,估计患病率为 1/11300-37600。大多数患者在数年无症状,直到腺瘤变大变多,导致直肠出血甚至贫血,或癌症发展。一般来说,癌症在息肉出现十年后开始发展。非特异性症状可能包括便秘或腹泻、腹痛、可触及的腹部肿块和体重减轻。FAP 可能会出现一些肠外表现,如骨瘤、牙齿异常(未萌出的牙齿、先天性缺失一颗或多颗牙齿、额外牙、齿龈囊肿和牙瘤)、视网膜色素上皮先天性肥大(CHRPE)、韧带样纤维瘤和结外癌症(甲状腺、肝脏、胆管和中枢神经系统)。FAP 的一种侵袭性较弱的变体,即衰减型 FAP(AFAP),其特征是结直肠腺瘤性息肉较少(通常为 10 至 100 个),腺瘤出现的年龄较晚,癌症风险较低。一些病变(颅骨和下颌骨骨瘤、牙齿异常、头皮、肩部、手臂和背部的纤维瘤)提示 Gardner 型 FAP。经典的 FAP 以常染色体显性方式遗传,是由腺瘤性息肉病(APC)基因的种系突变引起的。大多数患者(~70%)有结直肠息肉和癌症的家族史。在一部分个体中,MUTYH 突变导致隐性遗传的息肉病,即 MUTYH 相关息肉病(MAP),其特征是结直肠癌和上消化道和下消化道息肉/腺瘤的风险略有增加。诊断基于提示性家族史、临床发现和大肠内镜检查或全结肠镜检查。只要有可能,临床诊断应通过基因检测确认。当家族中的 APC 突变已确定时,应对所有一级亲属进行基因检测。可进行产前(羊膜穿刺术和绒毛膜活检)、甚至着床前(胚胎植入前)的遗传检测。必须转介给遗传学家或遗传咨询师。鉴别诊断包括其他引起多发性息肉的疾病(如 Peutz-Jeghers 综合征、家族性青少年息肉或增生性息肉、遗传性混合息肉综合征和 Lynch 综合征)。预防癌症和保持良好的生活质量是管理的主要目标,应向所有患者提供定期和系统的随访和支持性护理。在十几岁后期或二十岁出头时,提倡进行结直肠癌预防性手术。对于 AFAP,建议的替代方案是全直肠结肠切除术和回肠肛管吻合术或回肠直肠吻合术。十二指肠癌和韧带样纤维瘤是全结肠切除术后的两个主要死亡原因,需要早期识别并治疗。上消化道内镜检查是降低壶腹和十二指肠癌风险的必要手段。对于进展性肿瘤和无法切除的疾病,联合细胞毒性化疗和手术(如有可能进行)可能会有反应或稳定。在美国食品和药物管理局和欧洲药品管理局批准的 Celecoxib 辅助治疗 FAP 中。FAP 患者的结直肠癌风险为 100%;然而,当患者进入筛查-治疗计划时,这种风险会显著降低。