Choudhury Ananyo, Hazelhurst Scott, Meintjes Ayton, Achinike-Oduaran Ovokeraye, Aron Shaun, Gamieldien Junaid, Jalali Sefid Dashti Mahjoubeh, Mulder Nicola, Tiffin Nicki, Ramsay Michèle
Sydney Brenner Institute of Molecular Bioscience, University of the Witwatersrand, Johannesburg, South Africa.
BMC Genomics. 2014 Jun 6;15(1):437. doi: 10.1186/1471-2164-15-437.
Population differentiation is the result of demographic and evolutionary forces. Whole genome datasets from the 1000 Genomes Project (October 2012) provide an unbiased view of genetic variation across populations from Europe, Asia, Africa and the Americas. Common population-specific SNPs (MAF > 0.05) reflect a deep history and may have important consequences for health and wellbeing. Their interpretation is contextualised by currently available genome data.
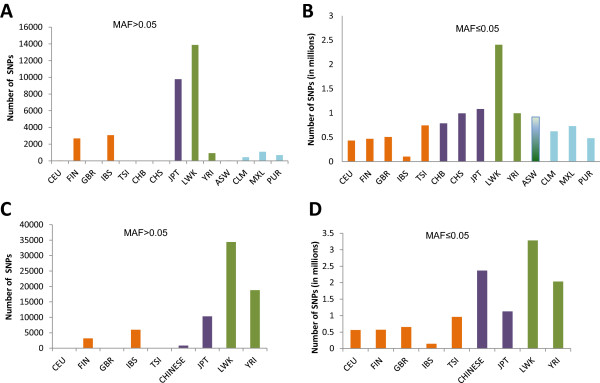
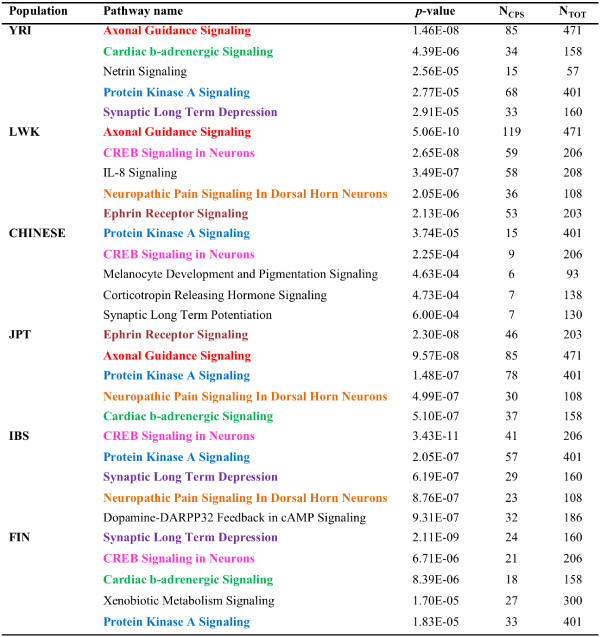
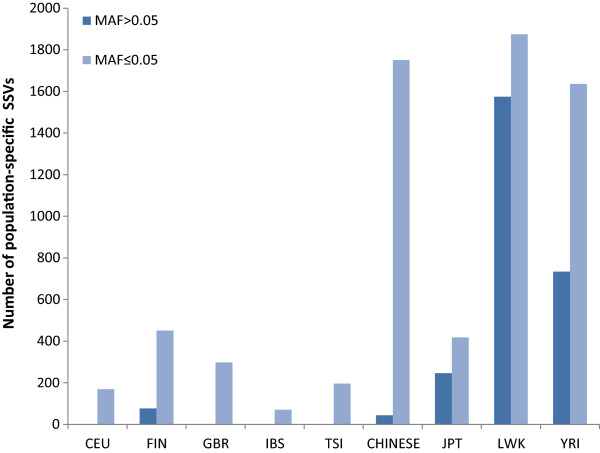
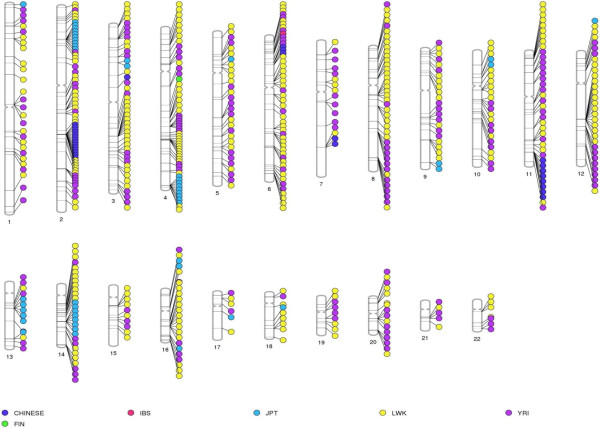
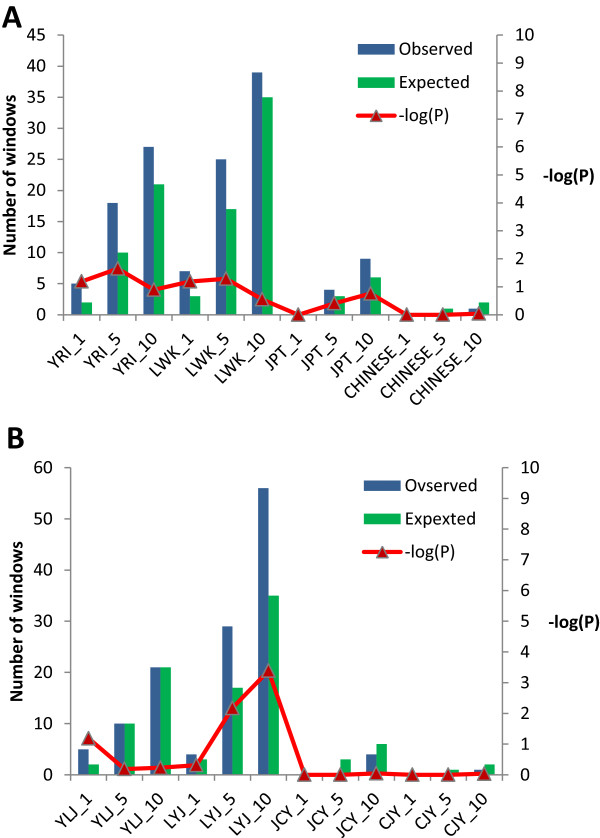
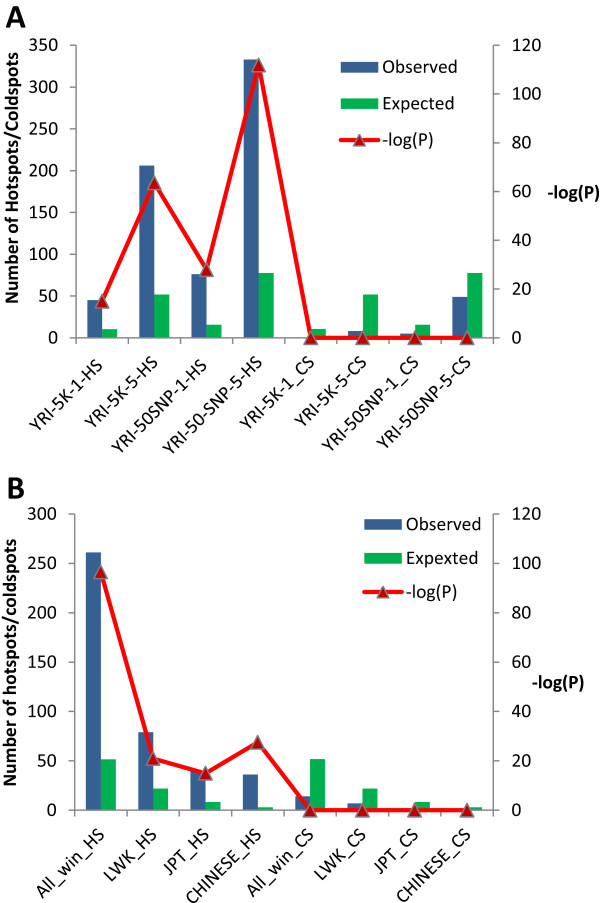
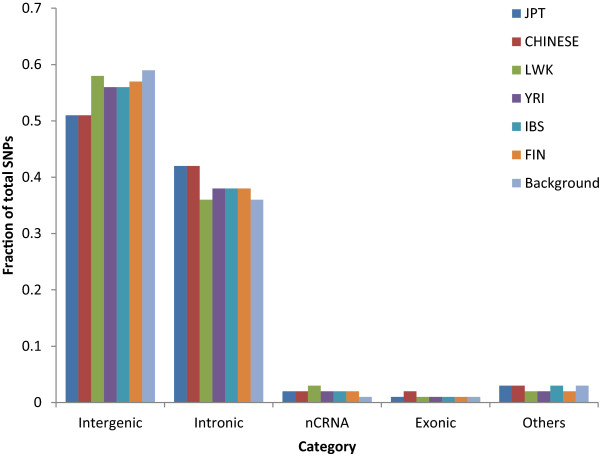
The identification of common population-specific (CPS) variants (SNPs and SSV) is influenced by admixture and the sample size under investigation. Nine of the populations in the 1000 Genomes Project (2 African, 2 Asian (including a merged Chinese group) and 5 European) revealed that the African populations (LWK and YRI), followed by the Japanese (JPT) have the highest number of CPS SNPs, in concordance with their histories and given the populations studied. Using two methods, sliding 50-SNP and 5-kb windows, the CPS SNPs showed distinct clustering across large genome segments and little overlap of clusters between populations. iHS enrichment score and the population branch statistic (PBS) analyses suggest that selective sweeps are unlikely to account for the clustering and population specificity. Of interest is the association of clusters close to recombination hotspots. Functional analysis of genes associated with the CPS SNPs revealed over-representation of genes in pathways associated with neuronal development, including axonal guidance signalling and CREB signalling in neurones.
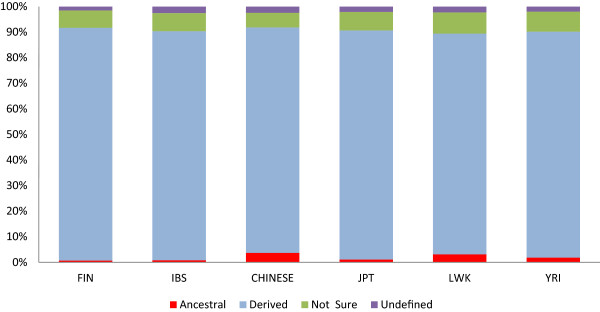
Common population-specific SNPs are non-randomly distributed throughout the genome and are significantly associated with recombination hotspots. Since the variant alleles of most CPS SNPs are the derived allele, they likely arose in the specific population after a split from a common ancestor. Their proximity to genes involved in specific pathways, including neuronal development, suggests evolutionary plasticity of selected genomic regions. Contrary to expectation, selective sweeps did not play a large role in the persistence of population-specific variation. This suggests a stochastic process towards population-specific variation which reflects demographic histories and may have some interesting implications for health and susceptibility to disease.
群体分化是人口统计学和进化力量作用的结果。来自千人基因组计划(2012年10月)的全基因组数据集为欧洲、亚洲、非洲和美洲群体的遗传变异提供了一个无偏差的视角。常见的群体特异性单核苷酸多态性(MAF > 0.05)反映了深远的历史,可能对健康和幸福有重要影响。它们的解释需结合当前可用的基因组数据。
常见群体特异性(CPS)变异(单核苷酸多态性和结构变异)的识别受混合情况和所研究样本量的影响。千人基因组计划中的9个群体(2个非洲群体、2个亚洲群体(包括合并的中国群体)和5个欧洲群体)显示,非洲群体(LWK和YRI),其次是日本群体(JPT),拥有数量最多的CPS单核苷酸多态性,这与其历史以及所研究的群体相符。使用两种方法,即滑动50个单核苷酸多态性和5千碱基窗口,CPS单核苷酸多态性在大的基因组片段上呈现出明显的聚类,且群体间聚类的重叠很少。iHS富集分数和群体分支统计(PBS)分析表明,选择性清除不太可能解释聚类和群体特异性。有趣的是靠近重组热点的聚类之间的关联。对与CPS单核苷酸多态性相关基因的功能分析显示,与神经元发育相关途径中的基因存在过度表达,包括轴突导向信号传导和神经元中的CREB信号传导。
常见的群体特异性单核苷酸多态性在整个基因组中呈非随机分布,且与重组热点显著相关。由于大多数CPS单核苷酸多态性的变异等位基因是衍生等位基因,它们可能在从共同祖先分裂后在特定群体中出现。它们与参与特定途径(包括神经元发育)的基因的接近性表明了所选基因组区域的进化可塑性。与预期相反,选择性清除在群体特异性变异的持续存在中并未起很大作用。这表明群体特异性变异是一个随机过程,反映了人口统计学历史,可能对健康和疾病易感性有一些有趣的影响。