The Microsoft Research-University of Trento, Centre for Computational Systems Biology, Povo (Trento), Italy.
BMC Bioinformatics. 2009 Nov 6;10:370. doi: 10.1186/1471-2105-10-370.
A great deal of data has accumulated on signalling pathways. These large datasets are thought to contain much implicit information on their molecular structure, interaction and activity information, which provides a picture of intricate molecular networks believed to underlie biological functions. While tremendous advances have been made in trying to understand these systems, how information is transmitted within them is still poorly understood. This ever growing amount of data demands we adopt powerful computational techniques that will play a pivotal role in the conversion of mined data to knowledge, and in elucidating the topological and functional properties of protein - protein interactions.
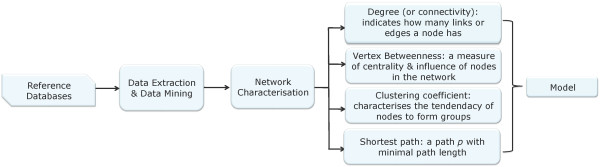
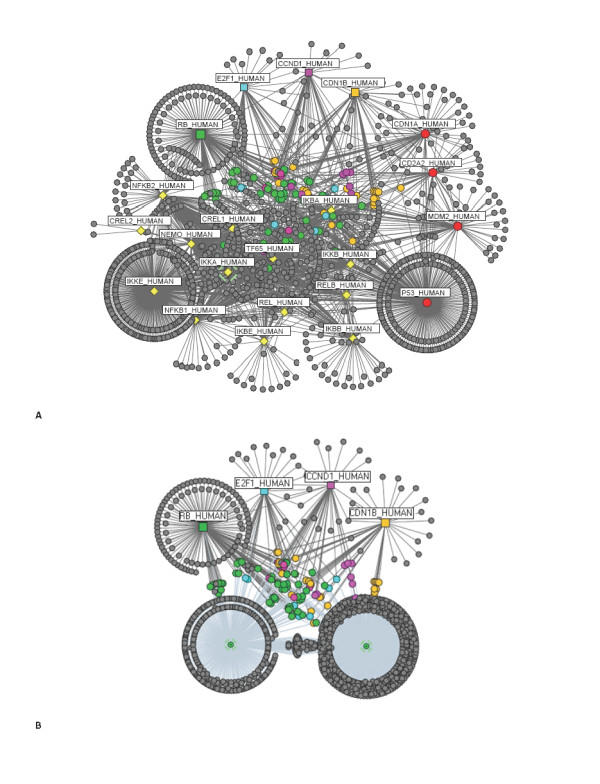
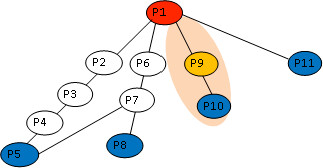
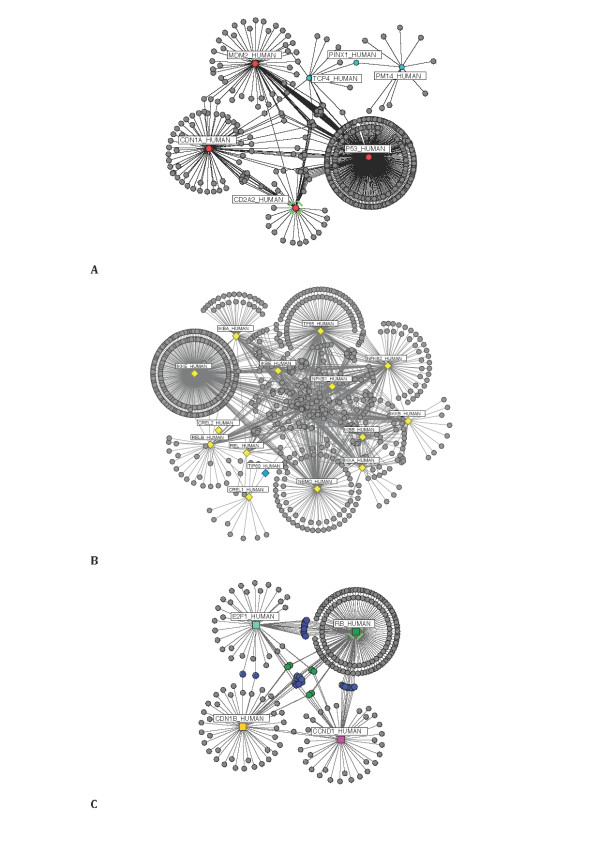
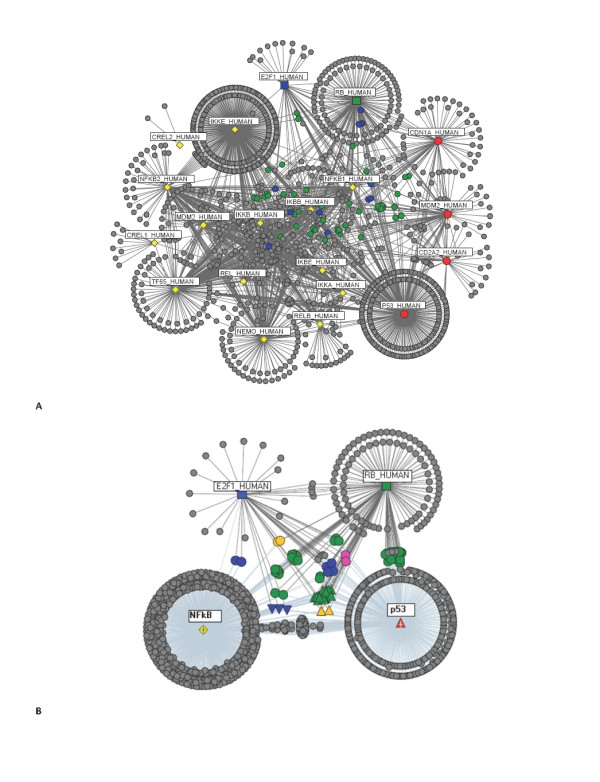
A computational framework is presented which allows for the description of embedded networks, and identification of common shared components thought to assist in the transmission of information within the systems studied. By employing the graph theories of network biology - such as degree distribution, clustering coefficient, vertex betweenness and shortest path measures - topological features of protein-protein interactions for published datasets of the p53, nuclear factor kappa B (NF-kappaB) and G1/S phase of the cell cycle systems were ascertained. Highly ranked nodes which in some cases were identified as connecting proteins most likely responsible for propagation of transduction signals across the networks were determined. The functional consequences of these nodes in the context of their network environment were also determined. These findings highlight the usefulness of the framework in identifying possible combination or links as targets for therapeutic responses; and put forward the idea of using retrieved knowledge on the shared components in constructing better organised and structured models of signalling networks.
It is hoped that through the data mined reconstructed signal transduction networks, well developed models of the published data can be built which in the end would guide the prediction of new targets based on the pathway's environment for further analysis. Source code is available upon request.
关于信号通路已经积累了大量的数据。这些大型数据集被认为包含了大量关于其分子结构、相互作用和活性信息的隐含信息,这些信息提供了一个复杂的分子网络的图景,这些网络被认为是生物功能的基础。尽管在试图理解这些系统方面已经取得了巨大的进展,但信息在这些系统内部是如何传递的仍然知之甚少。这种不断增长的数据量要求我们采用强大的计算技术,这些技术将在将挖掘到的数据转化为知识方面发挥关键作用,并阐明蛋白质-蛋白质相互作用的拓扑和功能特性。
提出了一种计算框架,允许描述嵌入式网络,并识别共同的共享组件,这些组件被认为有助于在研究系统中传递信息。通过运用网络生物学的图论,如度分布、聚类系数、顶点介数和最短路径度量,确定了已发表的 p53、核因子 kappa B(NF-kappaB)和细胞周期 G1/S 相系统的蛋白质-蛋白质相互作用的拓扑特征。确定了排名较高的节点,在某些情况下,这些节点被确定为连接蛋白,这些连接蛋白最有可能负责跨网络传播转导信号。还确定了这些节点在其网络环境中的功能后果。这些发现强调了该框架在识别可能的组合或链接作为治疗反应的目标方面的有用性;并提出了在构建信号网络的更好组织和结构化模型时利用共享组件的检索知识的想法。
希望通过挖掘重构的信号转导网络中的数据,可以构建出已发表数据的完善模型,最终将基于通路环境来预测新的靶点,以便进一步分析。可根据请求提供源代码。