Departments of Physiology and Medicine, Faculty of Medicine, University of Toronto, Toronto, Canada.
Diabetes. 2010 Feb;59(2):448-59. doi: 10.2337/db09-0129. Epub 2009 Nov 10.
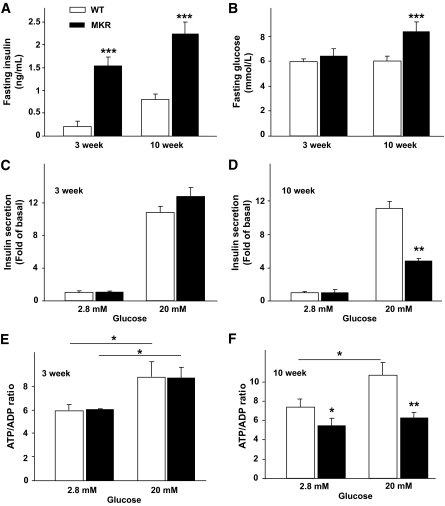
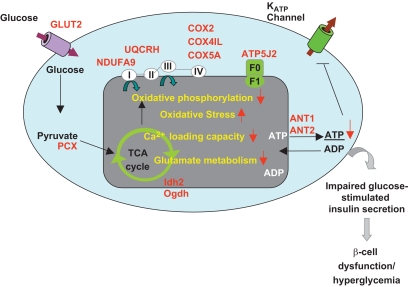
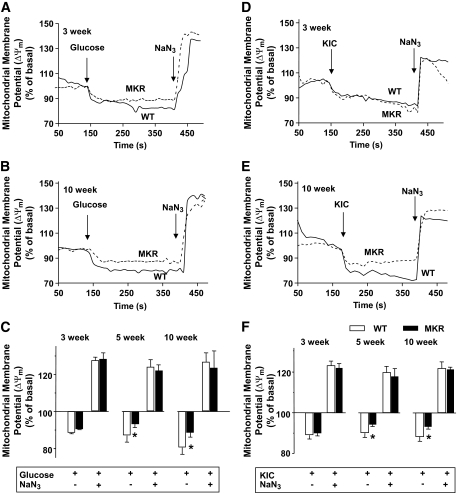
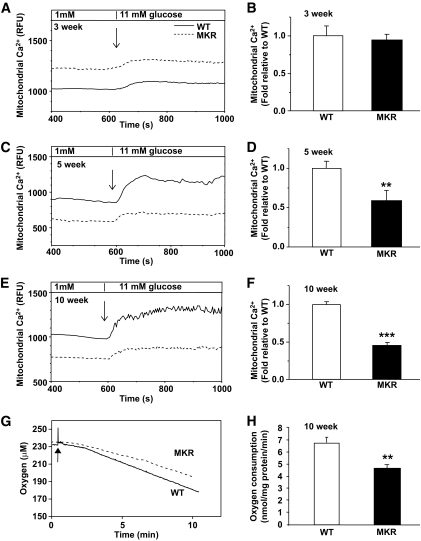
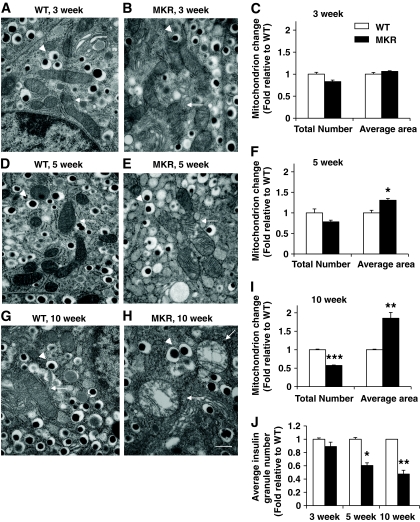
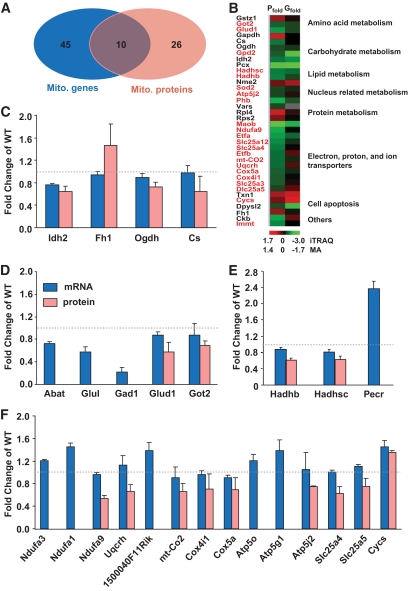
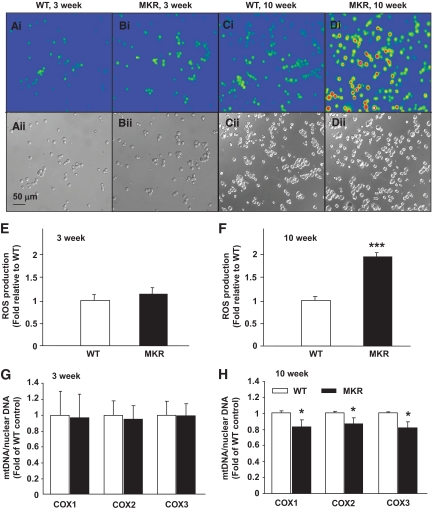
OBJECTIVE The inability of pancreatic beta-cells to appropriately respond to glucose and secrete insulin are primary defects associated with beta-cell failure in type 2 diabetes. Mitochondrial dysfunction has been implicated as a key factor in the development of type 2 diabetes; however, a link between mitochondrial dysfunction and defective insulin secretion is unclear. RESEARCH DESIGN AND METHODS We investigated the changes in islet mitochondrial function and morphology during progression from insulin resistance (3 weeks old), immediately before hyperglycemia (5 weeks old), and after diabetes onset (10 weeks old) in transgenic MKR mice compared with controls. The molecular and protein changes at 10 weeks were determined using microarray and iTRAQ proteomic screens. RESULTS At 3 weeks, MKR mice were hyperinsulinemic but normoglycemic and beta-cells showed negligible mitochondrial or morphological changes. At 5 weeks, MKR islets displayed abrogated hyperpolarization of mitochondrial membrane potential (DeltaPsi(m)), reduced mitochondrial Ca(2+) uptake, slightly enlarged mitochondria, and reduced glucose-stimulated insulin secretion. By 10 weeks, MKR mice were hyperglycemic and hyperinsulinemic and beta-cells contained swollen mitochondria with disordered cristae. beta-Cells displayed impaired stimulus-secretion coupling including reduced hyperpolarization of DeltaPsi(m), impaired Ca(2+)-signaling, and reduced glucose-stimulated ATP/ADP and insulin release. Furthermore, decreased cytochrome c oxidase-dependent oxygen consumption and signs of oxidative stress were observed in diabetic islets. Protein profiling of diabetic islets revealed that 36 mitochondrial proteins were differentially expressed, including inner membrane proteins of the electron transport chain. CONCLUSIONS We provide novel evidence for a critical role of defective mitochondrial oxidative phosphorylation and morphology in the pathology of insulin resistance-induced beta-cell failure.
胰腺β细胞不能适当响应葡萄糖并分泌胰岛素是 2 型糖尿病中β细胞衰竭的主要缺陷。线粒体功能障碍已被认为是 2 型糖尿病发展的关键因素;然而,线粒体功能障碍与胰岛素分泌缺陷之间的联系尚不清楚。
我们研究了与对照相比,在胰岛素抵抗(3 周龄)、高血糖前(5 周龄)和糖尿病发病后(10 周龄)期间,MKR 小鼠胰岛线粒体功能和形态的变化。使用微阵列和 iTRAQ 蛋白质组学筛选在 10 周时确定了分子和蛋白质的变化。
在 3 周时,MKR 小鼠高胰岛素血症但血糖正常,β细胞显示出微不足道的线粒体或形态变化。在 5 周时,MKR 胰岛显示出线粒体膜电位(DeltaPsi(m))去极化的丧失、线粒体 Ca(2+)摄取减少、线粒体略微增大和葡萄糖刺激的胰岛素分泌减少。到 10 周时,MKR 小鼠血糖升高和高胰岛素血症,β细胞含有肿胀的线粒体,嵴排列紊乱。β细胞显示出受损的刺激-分泌偶联,包括 DeltaPsi(m)去极化减少、Ca(2+)信号受损以及葡萄糖刺激的 ATP/ADP 和胰岛素释放减少。此外,在糖尿病胰岛中观察到细胞色素 c 氧化酶依赖性耗氧量减少和氧化应激迹象。糖尿病胰岛的蛋白质谱分析显示,有 36 种线粒体蛋白表达差异,包括电子传递链的内膜蛋白。
我们提供了新的证据,证明了缺陷的线粒体氧化磷酸化和形态在胰岛素抵抗诱导的β细胞衰竭的发病机制中起关键作用。