Okamoto Shu-ichi, Pouladi Mahmoud A, Talantova Maria, Yao Dongdong, Xia Peng, Ehrnhoefer Dagmar E, Zaidi Rameez, Clemente Arjay, Kaul Marcus, Graham Rona K, Zhang Dongxian, Vincent Chen H-S, Tong Gary, Hayden Michael R, Lipton Stuart A
Center for Neuroscience, Aging and Stem Cell Research, Burnham Institute for Medical Research, La Jolla, California, USA.
Nat Med. 2009 Dec;15(12):1407-13. doi: 10.1038/nm.2056. Epub 2009 Nov 15.
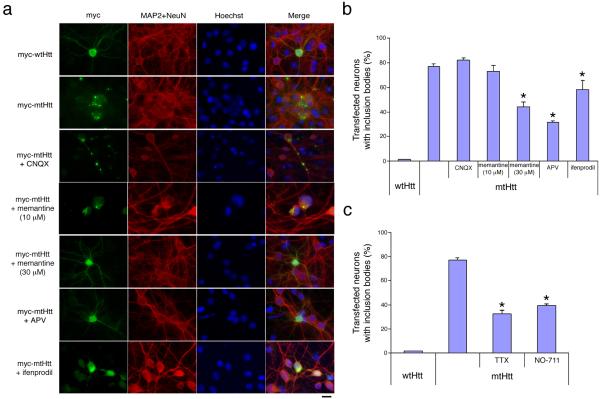
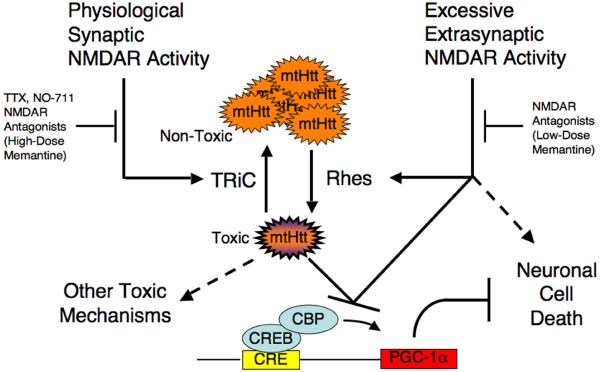
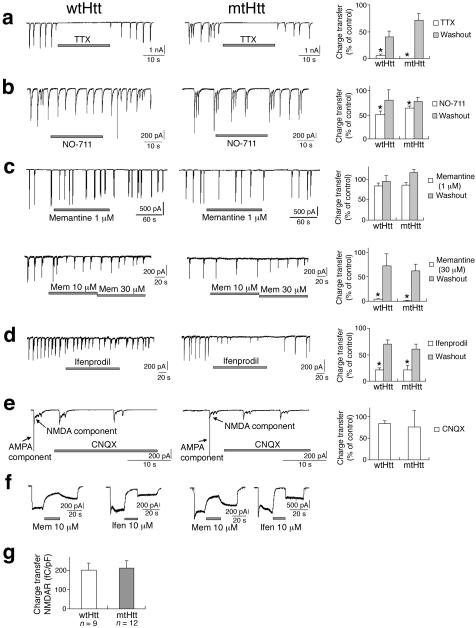
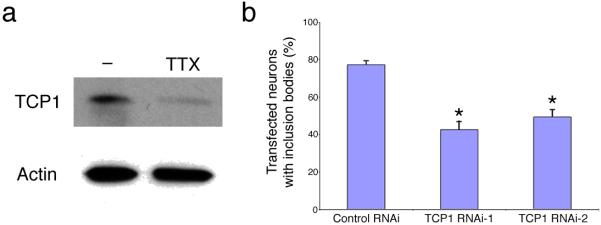
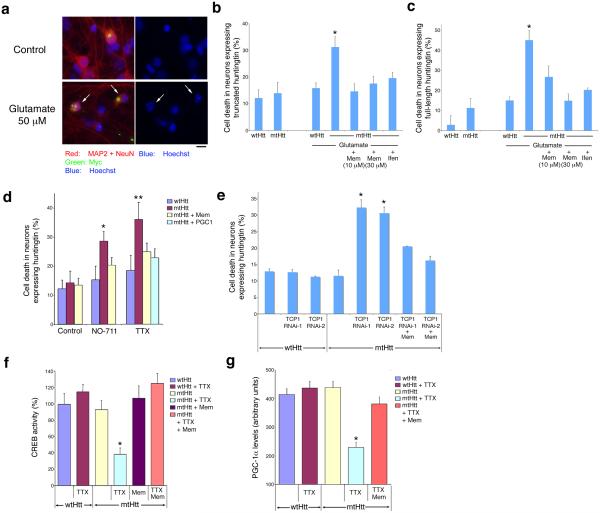
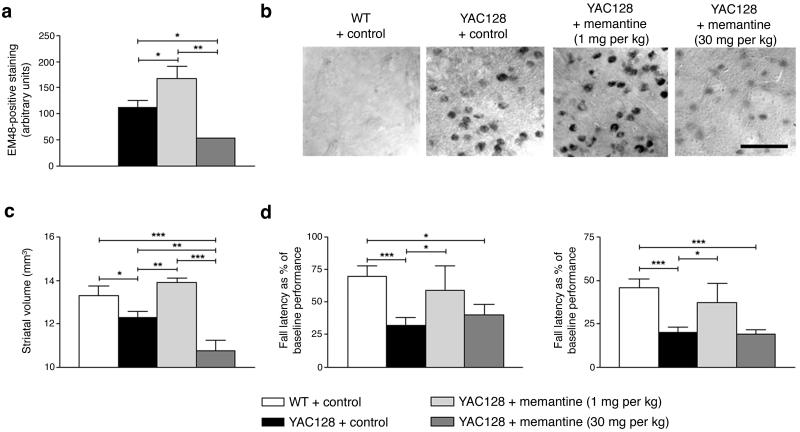
Huntington's disease is caused by an expanded CAG repeat in the gene encoding huntingtin (HTT), resulting in loss of striatal and cortical neurons. Given that the gene product is widely expressed, it remains unclear why neurons are selectively targeted. Here we show the relationship between synaptic and extrasynaptic activity, inclusion formation of mutant huntingtin protein (mtHtt) and neuronal survival. Synaptic N-methyl-D-aspartate-type glutamate receptor (NMDAR) activity induces mtHtt inclusions via a T complex-1 (TCP-1) ring complex (TRiC)-dependent mechanism, rendering neurons more resistant to mtHtt-mediated cell death. In contrast, stimulation of extrasynaptic NMDARs increases the vulnerability of mtHtt-containing neurons to cell death by impairing the neuroprotective cyclic AMP response element-binding protein (CREB)-peroxisome proliferator-activated receptor-gamma coactivator-1alpha (PGC-1alpha) cascade and increasing the level of the small guanine nucleotide-binding protein Rhes, which is known to sumoylate and disaggregate mtHtt. Treatment of transgenic mice expressing a yeast artificial chromosome containing 128 CAG repeats (YAC128) with low-dose memantine blocks extrasynaptic (but not synaptic) NMDARs and ameliorates neuropathological and behavioral manifestations. By contrast, high-dose memantine, which blocks both extrasynaptic and synaptic NMDAR activity, decreases neuronal inclusions and worsens these outcomes. Our findings offer a rational therapeutic approach for protecting susceptible neurons in Huntington's disease.
亨廷顿舞蹈症由编码亨廷顿蛋白(HTT)的基因中CAG重复序列扩增引起,导致纹状体和皮质神经元丧失。鉴于该基因产物广泛表达,尚不清楚为何神经元会被选择性靶向。在此,我们展示了突触和突触外活动、突变型亨廷顿蛋白(mtHtt)包涵体形成与神经元存活之间的关系。突触N-甲基-D-天冬氨酸型谷氨酸受体(NMDAR)活性通过T复合体-1(TCP-1)环复合体(TRiC)依赖性机制诱导mtHtt包涵体形成,使神经元对mtHtt介导的细胞死亡更具抗性。相反,突触外NMDAR的刺激会通过损害神经保护性环磷酸腺苷反应元件结合蛋白(CREB)-过氧化物酶体增殖物激活受体-γ共激活因子-1α(PGC-1α)级联反应并增加小GTP结合蛋白Rhes的水平,从而增加含mtHtt神经元对细胞死亡的易感性,已知Rhes会使mtHtt发生SUMO化并使其解聚。用低剂量美金刚治疗表达含128个CAG重复序列的酵母人工染色体(YAC128)的转基因小鼠,可阻断突触外(而非突触)NMDAR,并改善神经病理学和行为表现。相比之下,高剂量美金刚会阻断突触外和突触NMDAR活性,减少神经元包涵体并使这些结果恶化。我们的研究结果为保护亨廷顿舞蹈症中易感神经元提供了一种合理的治疗方法。