Center for Biosystems Research, University of Maryland Biotechnology Institute, College Park, 20742, USA.
BMC Evol Biol. 2009 Dec 2;9:280. doi: 10.1186/1471-2148-9-280.
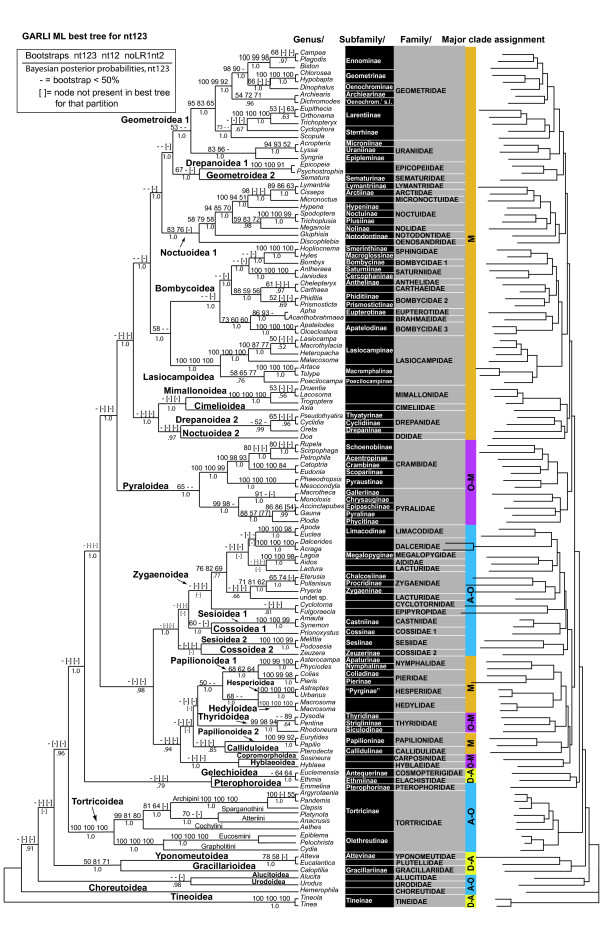
In the mega-diverse insect order Lepidoptera (butterflies and moths; 165,000 described species), deeper relationships are little understood within the clade Ditrysia, to which 98% of the species belong. To begin addressing this problem, we tested the ability of five protein-coding nuclear genes (6.7 kb total), and character subsets therein, to resolve relationships among 123 species representing 27 (of 33) superfamilies and 55 (of 100) families of Ditrysia under maximum likelihood analysis.
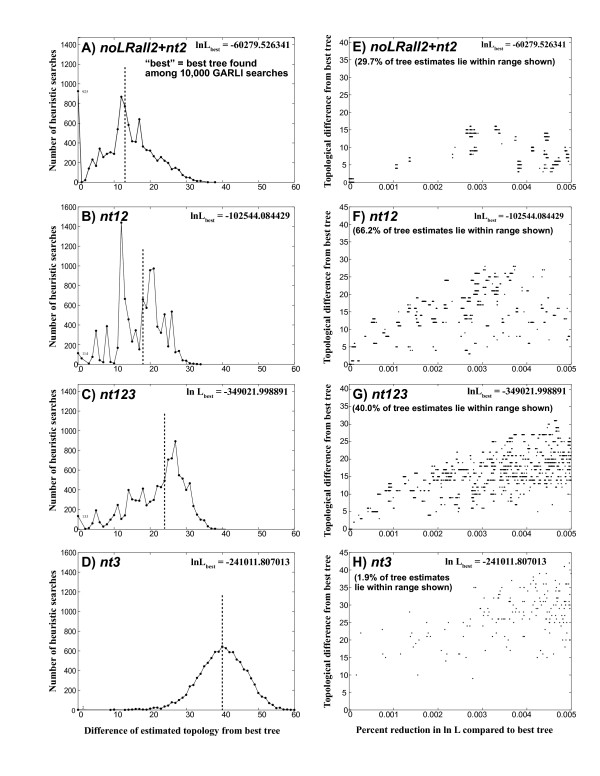
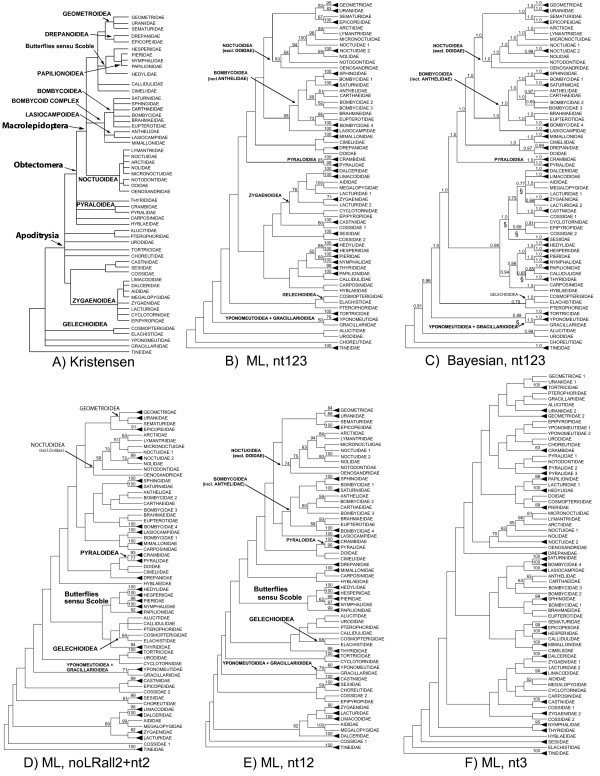
Our trees show broad concordance with previous morphological hypotheses of ditrysian phylogeny, although most relationships among superfamilies are weakly supported. There are also notable surprises, such as a consistently closer relationship of Pyraloidea than of butterflies to most Macrolepidoptera. Monophyly is significantly rejected by one or more character sets for the putative clades Macrolepidoptera as currently defined (P < 0.05) and Macrolepidoptera excluding Noctuoidea and Bombycoidea sensu lato (P < or = 0.005), and nearly so for the superfamily Drepanoidea as currently defined (P < 0.08). Superfamilies are typically recovered or nearly so, but usually without strong support. Relationships within superfamilies and families, however, are often robustly resolved. We provide some of the first strong molecular evidence on deeper splits within Pyraloidea, Tortricoidea, Geometroidea, Noctuoidea and others.Separate analyses of mostly synonymous versus non-synonymous character sets revealed notable differences (though not strong conflict), including a marked influence of compositional heterogeneity on apparent signal in the third codon position (nt3). As available model partitioning methods cannot correct for this variation, we assessed overall phylogeny resolution through separate examination of trees from each character set. Exploration of "tree space" with GARLI, using grid computing, showed that hundreds of searches are typically needed to find the best-feasible phylogeny estimate for these data.
Our results (a) corroborate the broad outlines of the current working phylogenetic hypothesis for Ditrysia, (b) demonstrate that some prominent features of that hypothesis, including the position of the butterflies, need revision, and (c) resolve the majority of family and subfamily relationships within superfamilies as thus far sampled. Much further gene and taxon sampling will be needed, however, to strongly resolve individual deeper nodes.
在鳞翅目(蝴蝶和蛾类;已描述的物种有 165000 种)这个种类繁多的昆虫目中,所属的双翅目(Ditrysia)内的较深层次关系了解甚少。为了解决这个问题,我们测试了五个编码核蛋白的基因(总长度为 6.7kb),以及其中的字符子集,以在最大似然分析中解决代表 Ditrysia 的 123 个物种(33 个超科中的 27 个和 100 个科中的 55 个)之间的关系。
我们的树与先前关于双翅目系统发育的形态学假说有广泛的一致性,尽管大多数超科之间的关系支持度较弱。也有一些显著的意外,例如与大多数鳞翅目相比,Pyraloidea 的亲缘关系更密切。一个或多个字符集显著拒绝当前定义的假定类群鳞翅目(Macrolepidoptera)(P<0.05)和不包括 Noctuoidea 和 Bombycoidea sensu lato 的鳞翅目(Macrolepidoptera)(P<0.005)的单系性,以及当前定义的 Drepanoidea 超科(P<0.08)几乎如此。超科通常被回收或几乎如此,但通常没有强烈的支持。然而,超科内和科内的关系通常得到很好的解决。我们提供了一些关于 Pyraloidea、Tortricoidea、Geometroidea、Noctuoidea 等内部更深层次分裂的第一个强有力的分子证据。对主要是同义与非同义字符集的单独分析显示出明显的差异(尽管不是强烈的冲突),包括组成异质性对第三密码子位置(nt3)中明显信号的显著影响。由于可用的模型分区方法无法纠正这种变化,我们通过分别检查每个字符集的树来评估总体系统发育分辨率。使用 Grid 计算的 GARLI 对“树空间”的探索表明,对于这些数据,通常需要数百次搜索才能找到最佳可行的系统发育估计值。
我们的结果(a)证实了双翅目当前工作系统发育假说的大致轮廓,(b)表明该假说的一些突出特征,包括蝴蝶的位置,需要修改,(c)解决了迄今为止采样的超科内大多数科和亚科关系。然而,需要进一步的基因和分类群采样,才能强烈解决单个更深层次的节点。