Center for Human Genetics Research, Department of Molecular Physiology and Biophysics, Vanderbilt University, Nashville, TN, USA.
BioData Min. 2009 Dec 3;2(1):7. doi: 10.1186/1756-0381-2-7.
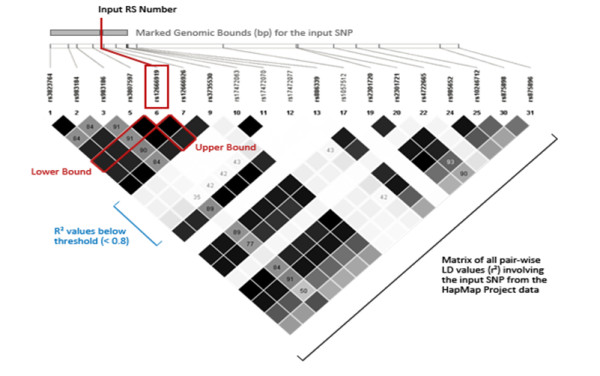
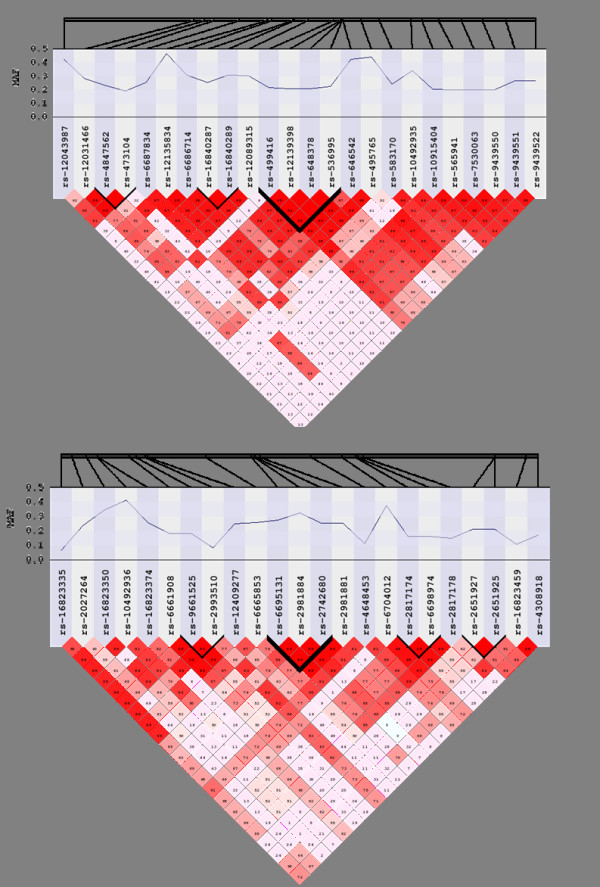
Gene-centric analysis tools for genome-wide association study data are being developed both to annotate single locus statistics and to prioritize or group single nucleotide polymorphisms (SNPs) prior to analysis. These approaches require knowledge about the relationships between SNPs on a genotyping platform and genes in the human genome. SNPs in the genome can represent broader genomic regions via linkage disequilibrium (LD), and population-specific patterns of LD can be exploited to generate a data-driven map of SNPs to genes.
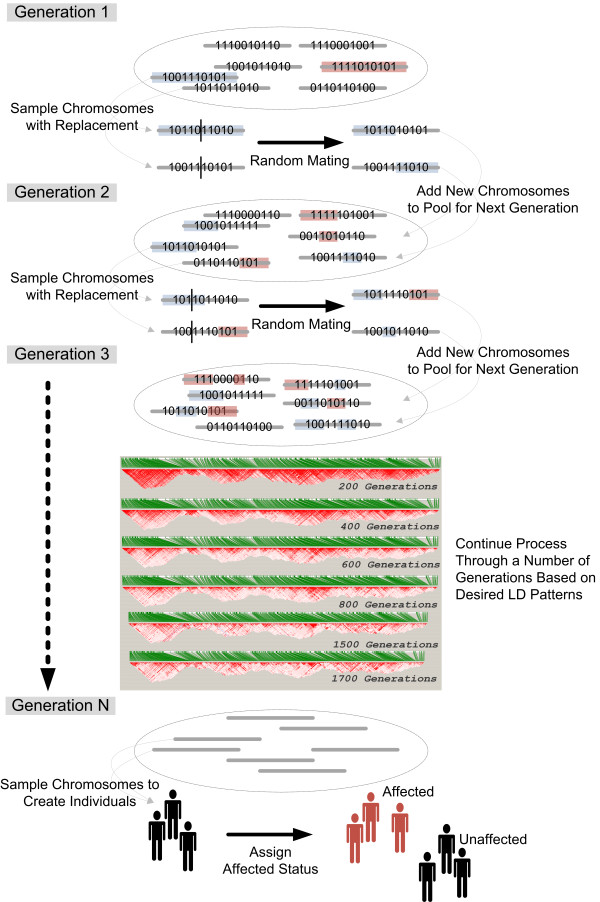
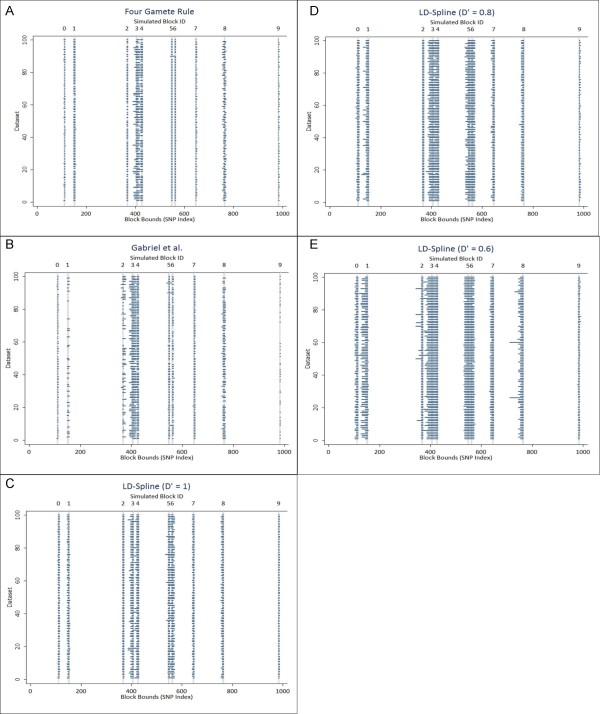
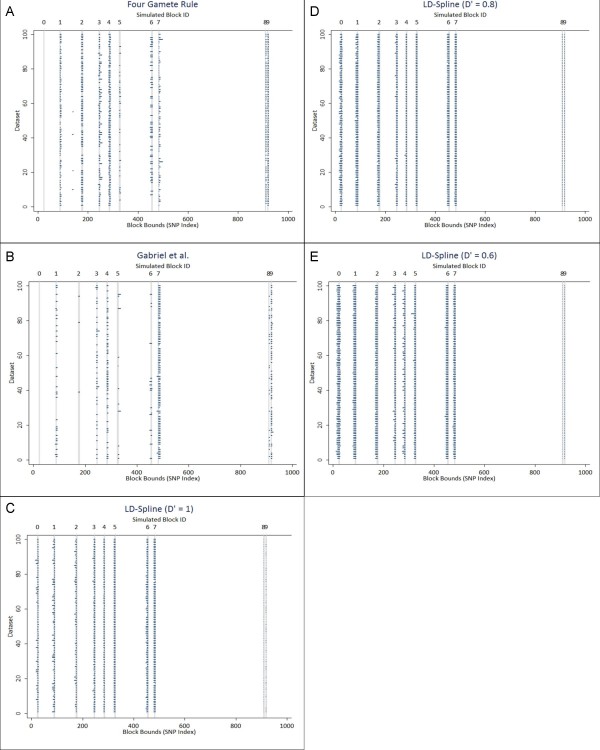
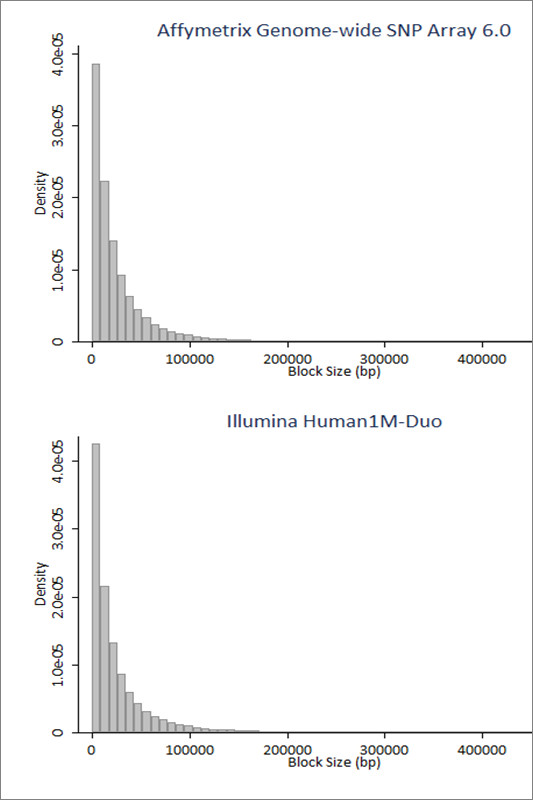
In this study, we implemented LD-Spline, a database routine that defines the genomic boundaries a particular SNP represents using linkage disequilibrium statistics from the International HapMap Project. We compared the LD-Spline haplotype block partitioning approach to that of the four gamete rule and the Gabriel et al. approach using simulated data; in addition, we processed two commonly used genome-wide association study platforms.
We illustrate that LD-Spline performs comparably to the four-gamete rule and the Gabriel et al. approach; however as a SNP-centric approach LD-Spline has the added benefit of systematically identifying a genomic boundary for each SNP, where the global block partitioning approaches may falter due to sampling variation in LD statistics.
LD-Spline is an integrated database routine that quickly and effectively defines the genomic region marked by a SNP using linkage disequilibrium, with a SNP-centric block definition algorithm.
全基因组关联研究数据的基因中心分析工具正在被开发,用于注释单基因座统计数据,并在分析前对单核苷酸多态性 (SNP) 进行优先级排序或分组。这些方法需要了解基因分型平台上 SNP 与人类基因组中基因之间的关系。基因组中的 SNP 可以通过连锁不平衡 (LD) 来代表更广泛的基因组区域,并且可以利用特定于群体的 LD 模式来生成基于数据的 SNP 到基因图谱。
在这项研究中,我们实现了 LD-Spline,这是一个数据库例程,它使用来自国际人类基因组单体型图计划的连锁不平衡统计信息来定义特定 SNP 所代表的基因组边界。我们比较了 LD-Spline 单倍型块划分方法与四配子规则和 Gabriel 等人的方法,使用模拟数据;此外,我们还处理了两个常用的全基因组关联研究平台。
我们说明 LD-Spline 的性能与四配子规则和 Gabriel 等人的方法相当;然而,作为一种 SNP 中心的方法,LD-Spline 具有系统地为每个 SNP 确定基因组边界的附加优势,而全局块划分方法可能由于 LD 统计数据的采样变化而失败。
LD-Spline 是一种集成的数据库例程,它使用连锁不平衡快速有效地定义 SNP 标记的基因组区域,具有 SNP 中心的块定义算法。