Department of Medicine, University of California San Diego, La Jolla, California, USA.
PLoS Comput Biol. 2009 Nov;5(11):e1000581. doi: 10.1371/journal.pcbi.1000581. Epub 2009 Nov 26.
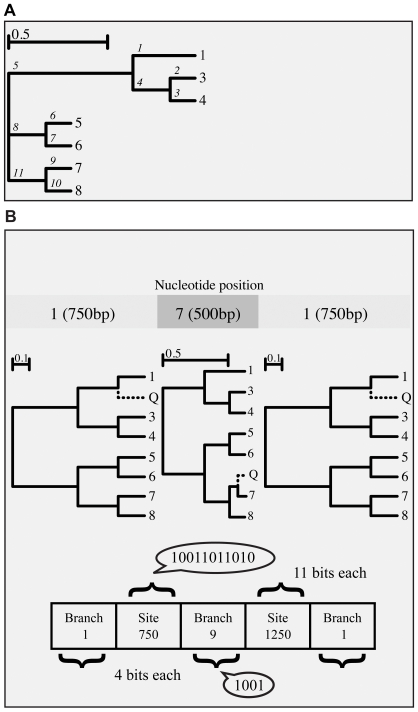
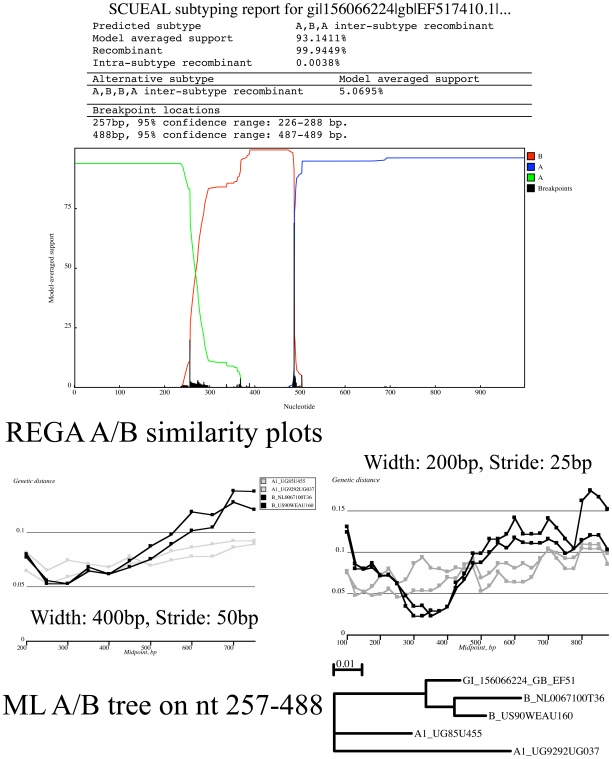
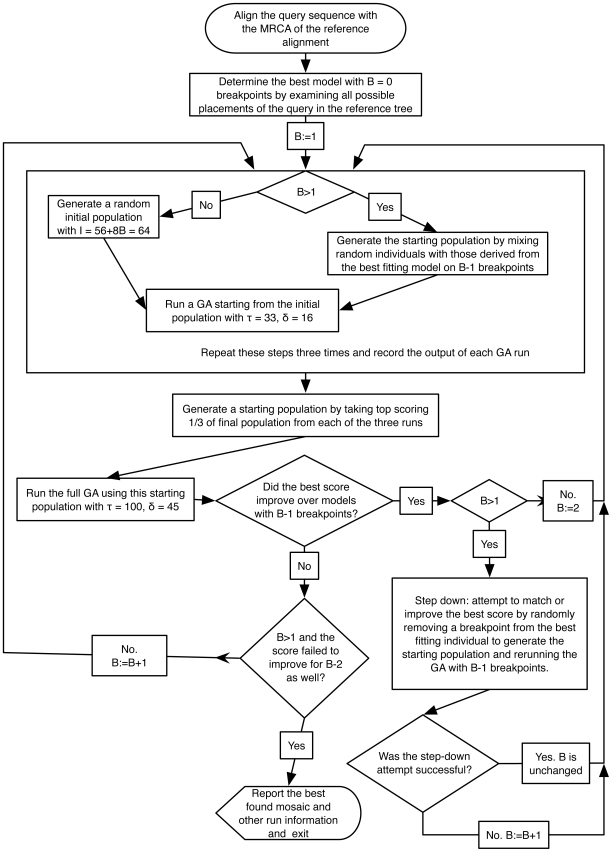
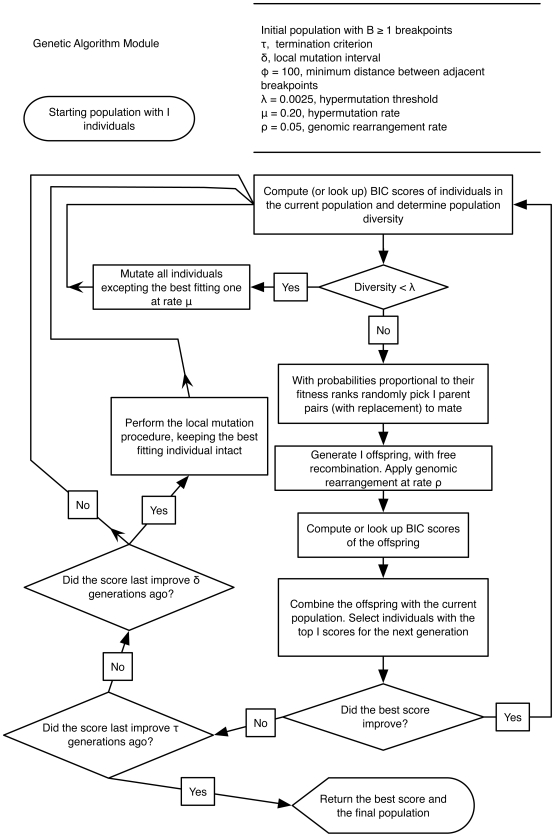
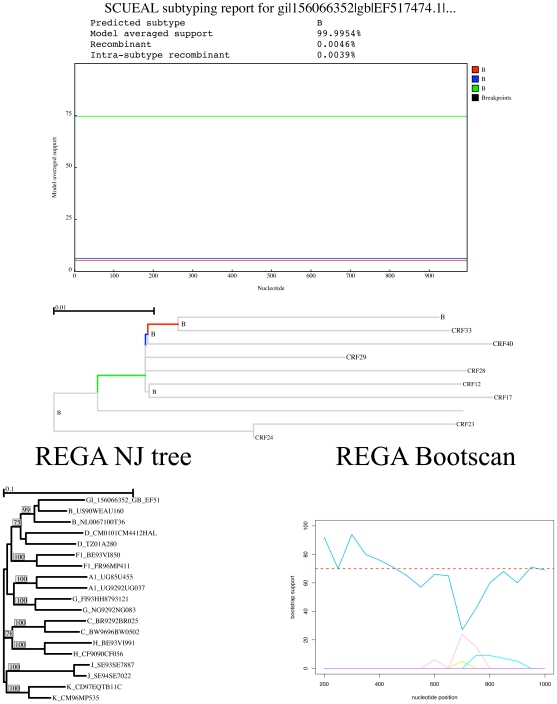
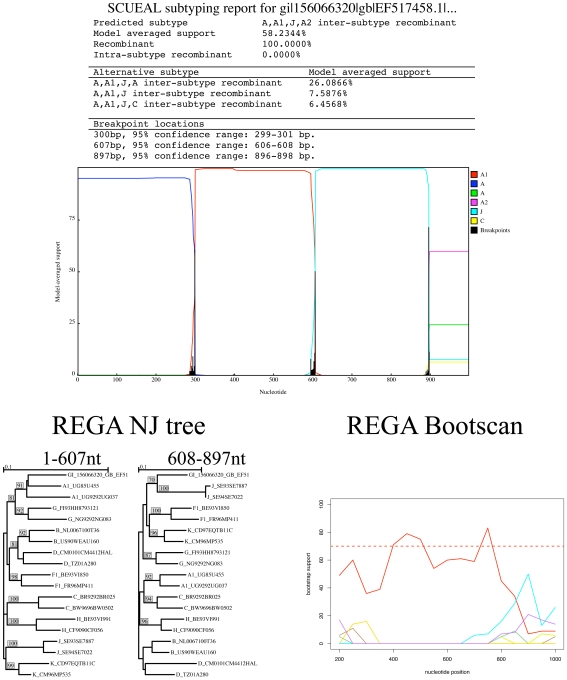
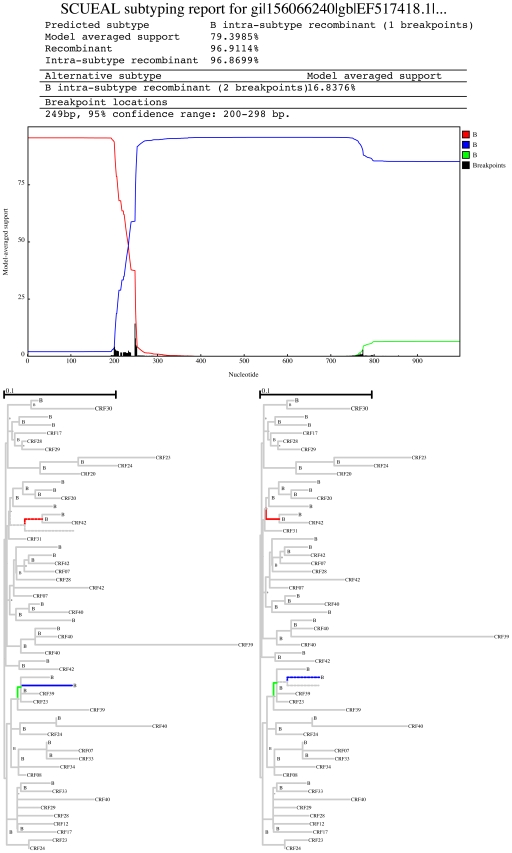
Genetically diverse pathogens (such as Human Immunodeficiency virus type 1, HIV-1) are frequently stratified into phylogenetically or immunologically defined subtypes for classification purposes. Computational identification of such subtypes is helpful in surveillance, epidemiological analysis and detection of novel variants, e.g., circulating recombinant forms in HIV-1. A number of conceptually and technically different techniques have been proposed for determining the subtype of a query sequence, but there is not a universally optimal approach. We present a model-based phylogenetic method for automatically subtyping an HIV-1 (or other viral or bacterial) sequence, mapping the location of breakpoints and assigning parental sequences in recombinant strains as well as computing confidence levels for the inferred quantities. Our Subtype Classification Using Evolutionary ALgorithms (SCUEAL) procedure is shown to perform very well in a variety of simulation scenarios, runs in parallel when multiple sequences are being screened, and matches or exceeds the performance of existing approaches on typical empirical cases. We applied SCUEAL to all available polymerase (pol) sequences from two large databases, the Stanford Drug Resistance database and the UK HIV Drug Resistance Database. Comparing with subtypes which had previously been assigned revealed that a minor but substantial (approximately 5%) fraction of pure subtype sequences may in fact be within- or inter-subtype recombinants. A free implementation of SCUEAL is provided as a module for the HyPhy package and the Datamonkey web server. Our method is especially useful when an accurate automatic classification of an unknown strain is desired, and is positioned to complement and extend faster but less accurate methods. Given the increasingly frequent use of HIV subtype information in studies focusing on the effect of subtype on treatment, clinical outcome, pathogenicity and vaccine design, the importance of accurate, robust and extensible subtyping procedures is clear.
遗传上多样化的病原体(如人类免疫缺陷病毒 1 型,HIV-1)经常根据系统发生或免疫定义的亚型进行分类。计算识别这些亚型有助于监测、流行病学分析和检测新的变体,例如 HIV-1 中的循环重组形式。已经提出了许多概念上和技术上不同的技术来确定查询序列的亚型,但没有一种方法是普遍最优的。我们提出了一种基于模型的系统发生方法,用于自动对 HIV-1(或其他病毒或细菌)序列进行亚型分类,确定断点的位置,并分配重组菌株中的亲本序列,以及计算推断数量的置信水平。我们的使用进化算法的亚型分类(SCUEAL)程序在各种模拟场景中表现非常出色,在筛选多个序列时可以并行运行,并且在典型的实际案例中可以与现有方法的性能相匹配或超过。我们将 SCUEAL 应用于来自两个大型数据库(斯坦福药物耐药性数据库和英国 HIV 药物耐药性数据库)的所有可用聚合酶(pol)序列。与以前分配的亚型进行比较表明,一小部分但相当大(约 5%)的纯亚型序列实际上可能是内部或亚型重组体。我们提供了一个免费的 SCUEAL 实现,作为 HyPhy 包和 Datamonkey 网络服务器的一个模块。当需要准确自动分类未知菌株时,我们的方法特别有用,并且可以补充和扩展更快但不太准确的方法。鉴于在关注亚型对治疗、临床结果、致病性和疫苗设计影响的研究中越来越频繁地使用 HIV 亚型信息,准确、稳健和可扩展的亚型分类程序的重要性显而易见。