Department of Zoology, North-Eastern Hill University, Shillong-793022, Meghalaya, India.
BMC Genomics. 2009 Dec 3;10 Suppl 3(Suppl 3):S25. doi: 10.1186/1471-2164-10-S3-S25.
Most phylogenetic studies using current methods have focused on primary DNA sequence information. However, RNA secondary structures are particularly useful in systematics because they include characteristics that give "morphological" information, not found in the primary sequence. In several mountainous regions of Northeastern India, foci of Paragonimus (lung fluke) infection reportedly involve species that are known to prevail in neighbouring countries. The present study was undertaken to demonstrate the sequence analysis of the ribosomal DNA (ITS2) of the infective (metacercarial) stage of the lung fluke collected from the edible crab hosts that are abundant in a mountain stream of the area (Miao, Changlang District in Arunachal Pradesh) and to construct its phylogeny. Using the approach of molecular morphometrics that is based on ITS2 secondary structure homologies, phylogenetic relationships of the various isolates of Paragonimus species that are prevalent in the neighbouring Near-eastern countries have been discussed.
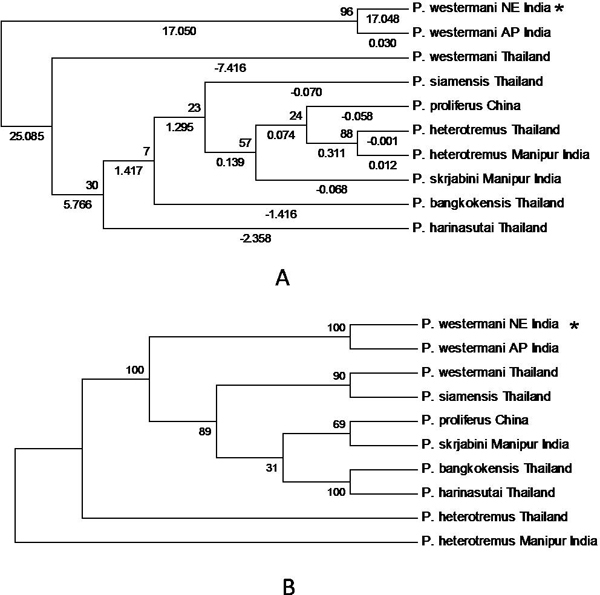
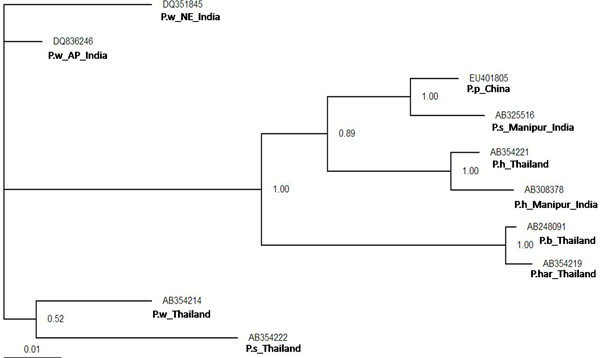
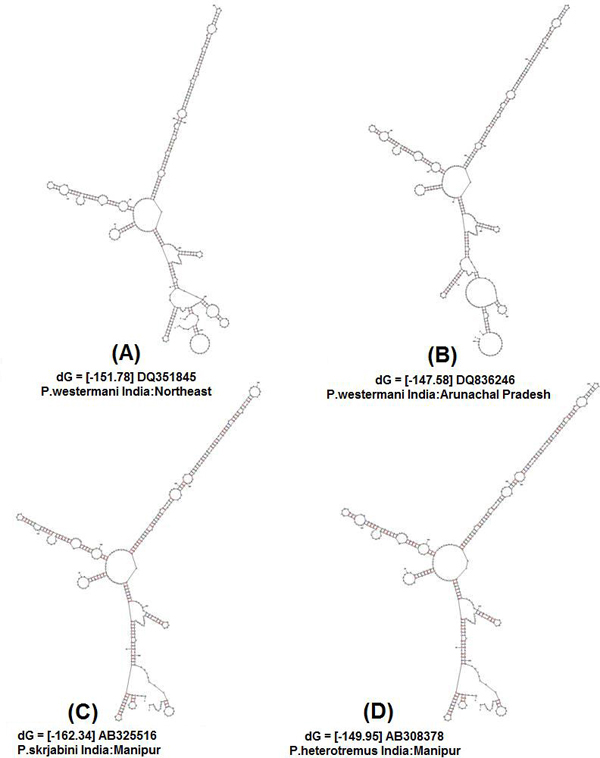
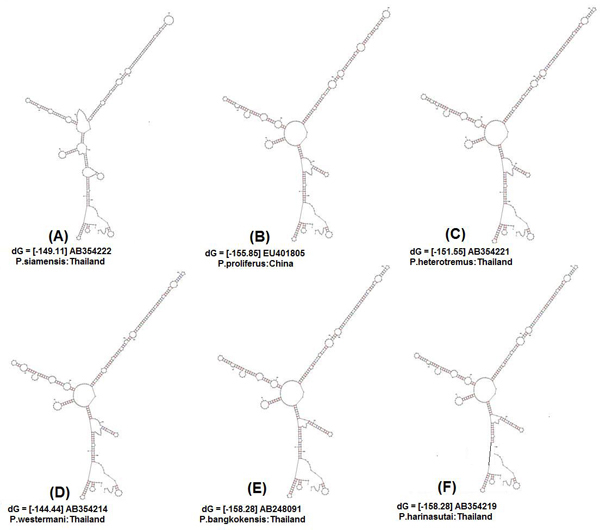
Initially, ten predicted RNA secondary structures were reconstructed and the topology based only on the predicted RNA secondary structure of the ITS2 region resolved most relationships among the species studied. We obtained three similar topologies for seven species of the genus Paragonimus on the basis of traditional primary sequence analysis using MEGA and a Bayesian analysis of the combined data. The latter approach allowed us to include both primary sequence and RNA molecular morphometrics; each data partition was allowed to have a different evolution rate. Paragonimus westermani was found to group with P. siamensis of Thailand; this was best supported by both the molecular morphometrics and combined analyses. P. heterotremus, P. proliferus, P. skrjabini, P. bangkokensis and P. harinasutai formed a separate clade in the molecular phylogenies, and were reciprocally monophyletic with respect to other species. ITS2 sequence motifs allowed an accurate in-silico distinction of lung flukes.
Data indicate that ITS2 motifs (<or= 50 bp in size) can be considered a promising tool for trematode species identification. RNA secondary structure analysis could be a valuable tool for distinguishing new species and completing Paragonimus systematics, more so because ITS2 secondary structure contains more information than the usual primary sequence alignment.
目前的大多数系统发育研究都集中在原始 DNA 序列信息上。然而,RNA 二级结构在系统学中特别有用,因为它们包含了在原始序列中找不到的“形态”特征。在印度东北部的几个山区,据报道有一些肺吸虫(肺部吸虫)感染的焦点涉及到在邻国流行的物种。本研究旨在展示从该地区(阿鲁纳恰尔邦昌朗区苗)一条山区溪流中丰富的食用蟹宿主中采集的肺吸虫感染(囊蚴)阶段的核糖体 DNA(ITS2)的序列分析,并构建其系统发育。利用基于 ITS2 二级结构同源性的分子形态计量学方法,讨论了在邻国流行的各种 Paragonimus 物种的分离株的系统发育关系。
最初,重建了十个预测的 RNA 二级结构,仅基于 ITS2 区域预测的 RNA 二级结构的拓扑解决了所研究物种之间的大多数关系。我们基于传统的主要序列分析,使用 MEGA 和联合数据的贝叶斯分析,获得了七种 Paragonimus 物种的三个相似拓扑。后一种方法允许我们包括主要序列和 RNA 分子形态计量学;允许每个数据分区具有不同的进化率。Paragonimus westermani 与泰国的 P. siamensis 聚在一起;这两种方法都得到了分子形态计量学和联合分析的最佳支持。P. heterotremus、P. proliferus、P. skrjabini、P. bangkokensis 和 P. harinasutai 在分子系统发育中形成了一个单独的分支,并且相对于其他物种具有互惠的单系性。ITS2 序列基序允许在计算机上准确区分肺吸虫。
数据表明,ITS2 基序(大小<=50bp)可被视为一种有前途的吸虫种鉴定工具。RNA 二级结构分析可能是区分新物种和完善 Paragonimus 系统发育的一种有价值的工具,因为 ITS2 二级结构比通常的原始序列比对包含更多的信息。