Department of Pharmacology, University of Firenze, Firenze, Italy.
BMC Genomics. 2009 Dec 11;10:596. doi: 10.1186/1471-2164-10-596.
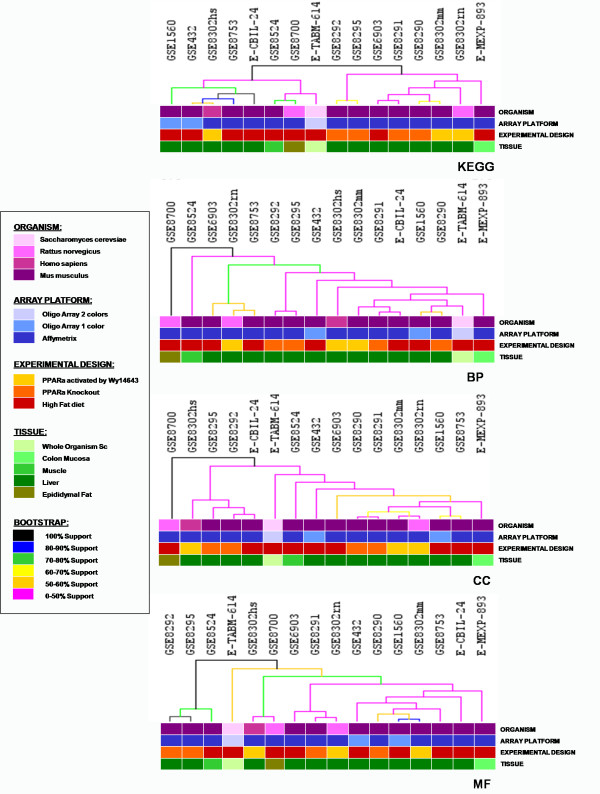
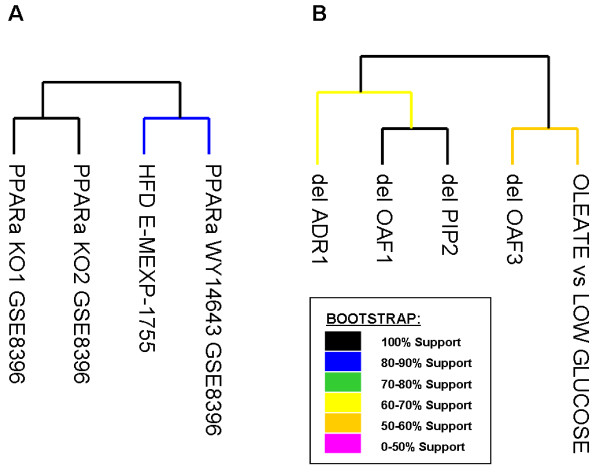
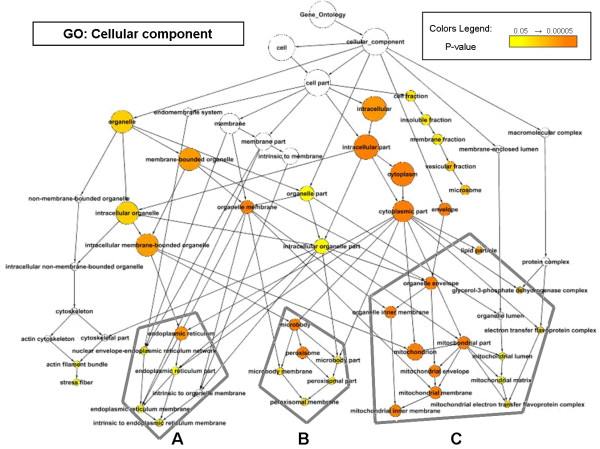
The application of high-throughput genomic tools in nutrition research is a widespread practice. However, it is becoming increasingly clear that the outcome of individual expression studies is insufficient for the comprehensive understanding of such a complex field. Currently, the availability of the large amounts of expression data in public repositories has opened up new challenges on microarray data analyses. We have focused on PPARalpha, a ligand-activated transcription factor functioning as fatty acid sensor controlling the gene expression regulation of a large set of genes in various metabolic organs such as liver, small intestine or heart. The function of PPARalpha is strictly connected to the function of its target genes and, although many of these have already been identified, major elements of its physiological function remain to be uncovered. To further investigate the function of PPARalpha, we have applied a cross-species meta-analysis approach to integrate sixteen microarray datasets studying high fat diet and PPARalpha signal perturbations in different organisms.
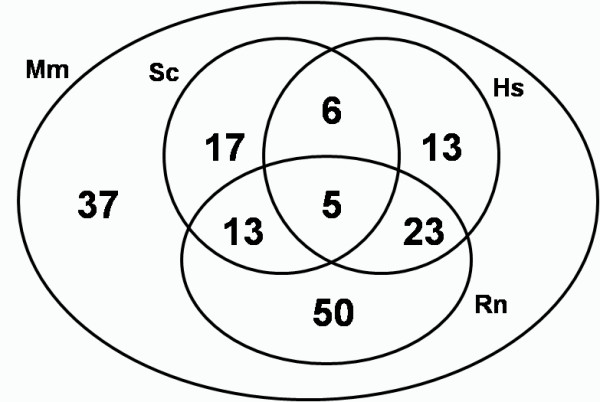
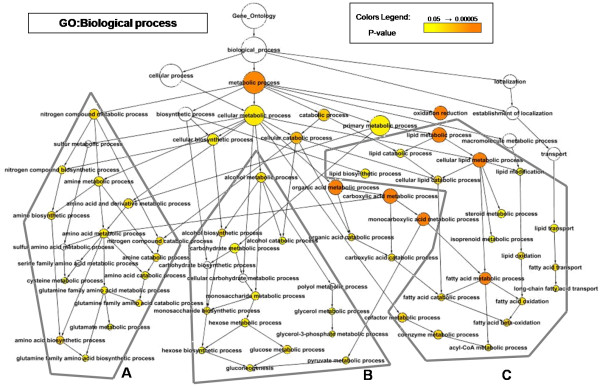
We identified 164 genes (MDEGs) that were differentially expressed in a constant way in response to a high fat diet or to perturbations in PPARs signalling. In particular, we found five genes in yeast which were highly conserved and homologous of PPARalpha targets in mammals, potential candidates to be used as models for the equivalent mammalian genes. Moreover, a screening of the MDEGs for all known transcription factor binding sites and the comparison with a human genome-wide screening of Peroxisome Proliferating Response Elements (PPRE), enabled us to identify, 20 new potential candidate genes that show, both binding site, both change in expression in the condition studied. Lastly, we found a non random localization of the differentially expressed genes in the genome.
The results presented are potentially of great interest to resume the currently available expression data, exploiting the power of in silico analysis filtered by evolutionary conservation. The analysis enabled us to indicate potential gene candidates that could fill in the gaps with regards to the signalling of PPARalpha and, moreover, the non-random localization of the differentially expressed genes in the genome, suggest that epigenetic mechanisms are of importance in the regulation of the transcription operated by PPARalpha.
高通量基因组工具在营养研究中的应用已十分普遍。然而,越来越明显的是,个体表达研究的结果不足以全面理解如此复杂的领域。目前,公共存储库中大量表达数据的可用性为微阵列数据分析带来了新的挑战。我们专注于过氧化物酶体增殖物激活受体α(PPARalpha),这是一种配体激活的转录因子,作为脂肪酸传感器,控制着肝脏、小肠或心脏等各种代谢器官中一大组基因的基因表达调控。PPARalpha 的功能与它的靶基因的功能密切相关,尽管其中许多已经被识别,但它的生理功能的主要元素仍有待发现。为了进一步研究 PPARalpha 的功能,我们应用了跨物种荟萃分析方法,整合了 16 个研究不同生物体中高脂肪饮食和 PPARalpha 信号扰动对微阵列数据集的影响。
我们确定了 164 个基因(MDEGs),它们对高脂肪饮食或 PPAR 信号扰动的反应方式一致。特别地,我们在酵母中发现了 5 个基因,它们与哺乳动物中 PPARalpha 靶标高度保守和同源,是作为哺乳动物等效基因模型的潜在候选基因。此外,对 MDEGs 进行所有已知转录因子结合位点的筛选,并与人类全基因组过氧化物酶体增殖物反应元件(PPRE)筛选进行比较,使我们能够识别出 20 个新的潜在候选基因,这些基因既具有结合位点,又在研究条件下改变表达。最后,我们发现差异表达基因在基因组中的定位是非随机的。
这些结果具有很大的研究潜力,可以利用进化保守性筛选过滤的计算分析的力量,总结当前可用的表达数据。该分析使我们能够指出潜在的基因候选者,这些候选者可以填补关于 PPARalpha 信号的空白,此外,差异表达基因在基因组中的非随机定位表明,表观遗传机制在 PPARalpha 调控的转录中很重要。