Department of Pharmacology, Weill Cornell Medical College, New York, NY 10065, USA Department of Neurology and the Pain and Palliative Care Service, Memorial Sloan-Kettering Cancer Center, 1275 York Avenue, New York, NY 10065, USA.
Pain. 2010 Feb;148(2):237-246. doi: 10.1016/j.pain.2009.11.003. Epub 2009 Dec 11.
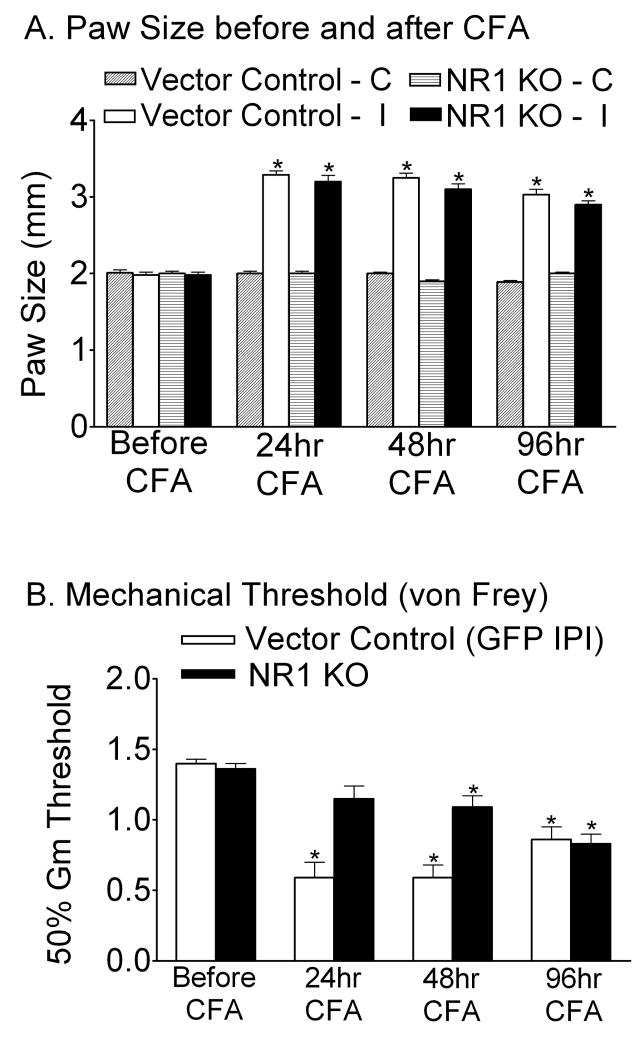
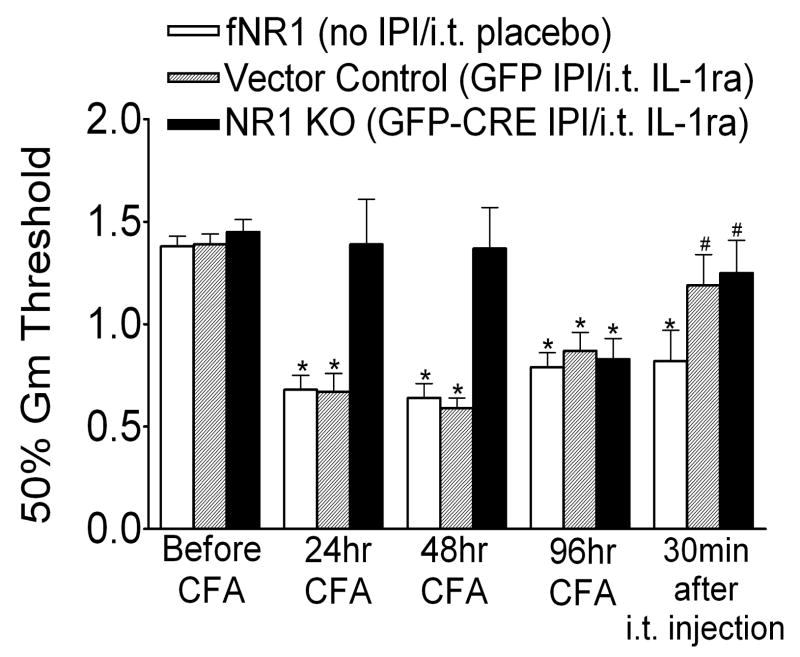
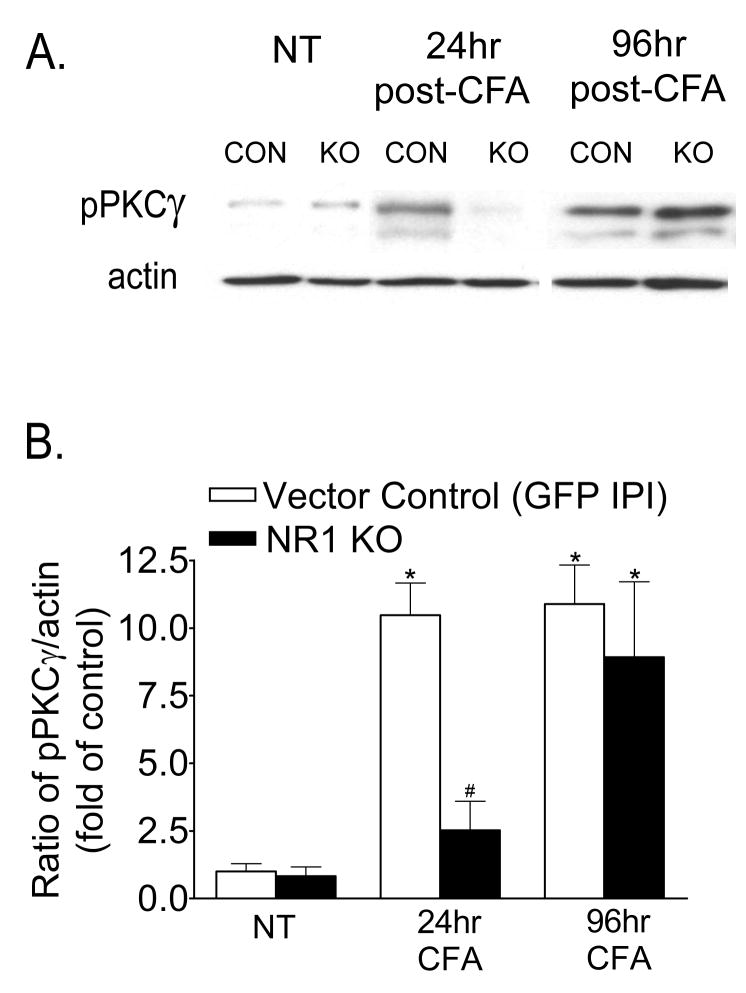
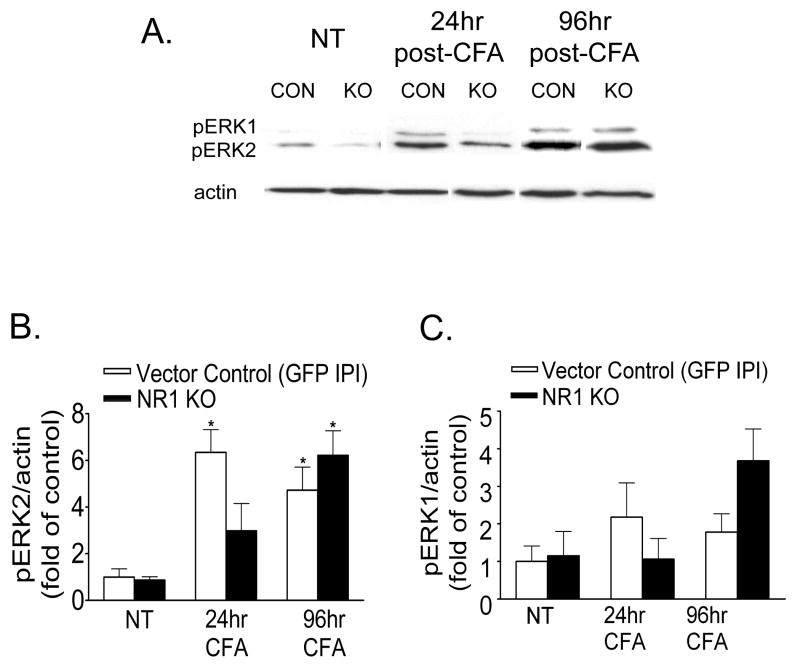
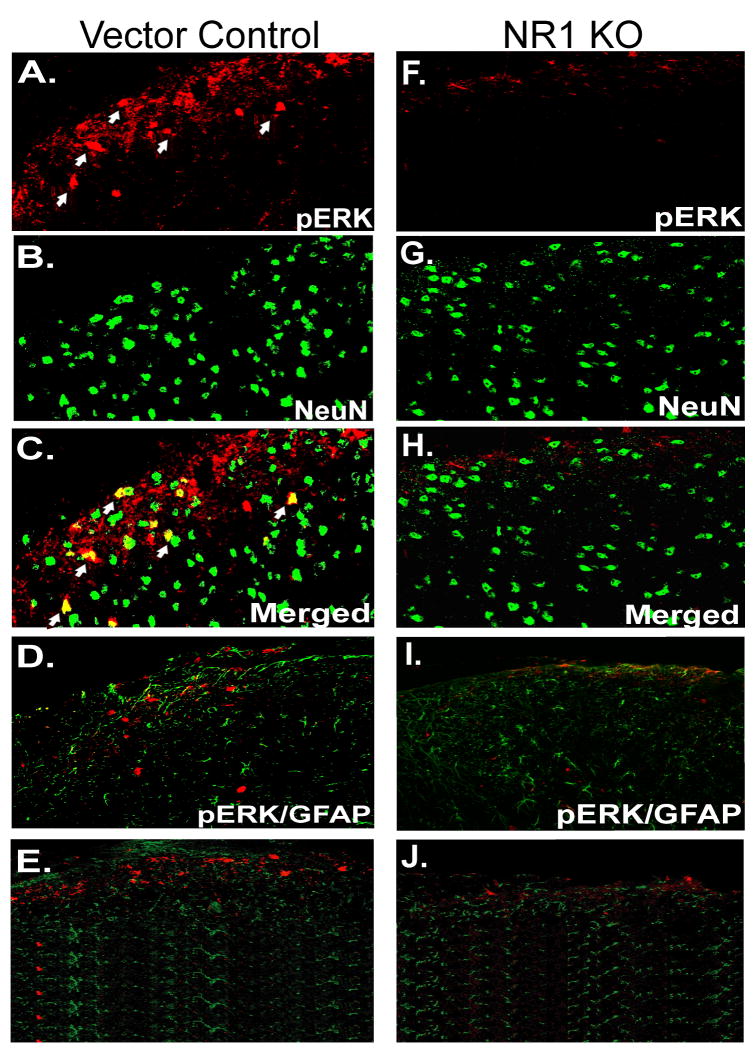
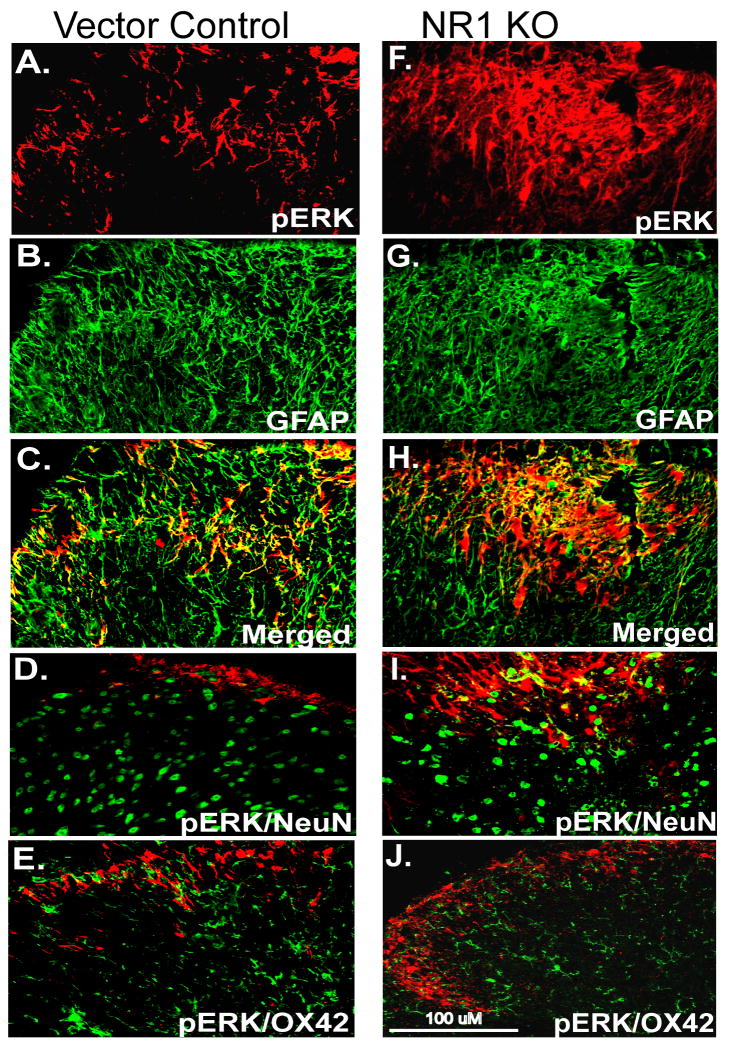
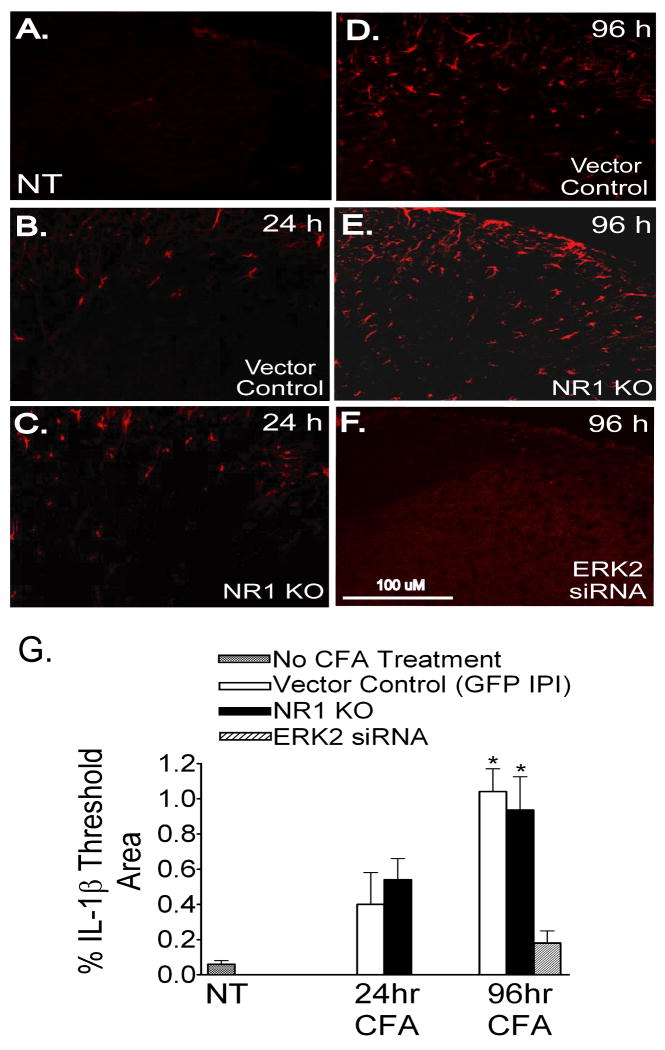
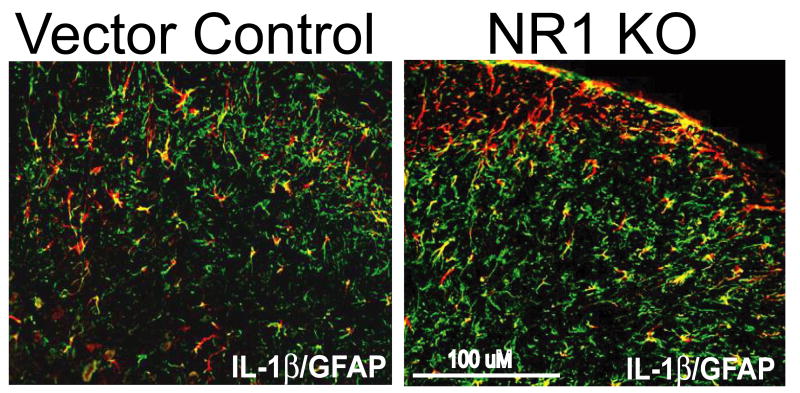
Following peripheral inflammation, NMDA receptor (NMDAR) activation in spinal cord dorsal horn neurons facilitates the generation of pain in response to low threshold inputs (allodynia) and signals the phosphorylation of protein kinase C (pPKC) and extracellular signal-regulated kinase 2 (pERK2). Intraplantar complete Freund's adjuvant (CFA) induces inflammatory nociception (allodynic pain) at 24 hours (h) with a concurrent increase in neuronal pPKCgamma and pERK2 but not glial pERK2. These effects are attenuated in a spatial knockout of the NMDAR (NR1 KO) confined to SCDH neurons. Although glia and proinflammatory cytokines are implicated in the maintenance of inflammatory pain and neuronal activation, the role of NMDARs and neuronal-glial-cytokine interactions that initiate and maintain inflammatory pain are not well defined. In the maintenance phase of inflammatory pain at 96h after CFA the NR1 KO mice are no longer protected from allodynia and the SCDH expression of pPKCgamma and pERK2 are increased. At 96h the expression of the proinflammatory cytokine, IL-1beta, and pERK2 are increased in astrocytes. Intrathecal IL-1 receptor antagonist (IL-1ra), acting on neuronal IL-1 receptors, completely reverses the allodynia at 96h after CFA. Deletion of NMDAR-dependent signaling in neurons protects against early CFA-induced allodynia. Subsequent NMDAR-independent signaling that involves neuronal expression of pPKCgamma and the induction of pERK2 and IL-1beta in activated astrocytes contributes to the emergence of NMDAR-independent inflammatory pain behavior at 96h after CFA. Effective reduction of the initiation and maintenance of inflammatory pain requires targeting the neuron-astrocyte-cytokine interactions revealed in these studies.
外周炎症后,脊髓背角神经元中的 NMDA 受体(NMDAR)激活促进了对低阈值输入(痛觉过敏)的疼痛产生,并信号转导蛋白激酶 C(pPKC)和细胞外信号调节激酶 2(pERK2)的磷酸化。足底完全弗氏佐剂(CFA)在 24 小时(h)内诱导炎症性伤害感受(痛觉过敏),同时神经元 pPKCγ和 pERK2增加,但胶质细胞 pERK2 不增加。这些影响在空间敲除脊髓背角神经元中的 NMDAR(NR1 KO)中减弱。尽管神经胶质细胞和促炎细胞因子参与炎症性疼痛的维持和神经元激活,但 NMDAR 以及启动和维持炎症性疼痛的神经元-神经胶质-细胞因子相互作用的作用尚未明确。在 CFA 后 96 小时炎症性疼痛的维持阶段,NR1 KO 小鼠不再免受痛觉过敏的影响,脊髓背角神经元 pPKCγ和 pERK2 的表达增加。在 96 小时时,星形胶质细胞中促炎细胞因子 IL-1β和 pERK2 的表达增加。鞘内注射白细胞介素-1 受体拮抗剂(IL-1ra),作用于神经元 IL-1 受体,可完全逆转 CFA 后 96 小时的痛觉过敏。神经元中 NMDAR 依赖性信号的缺失可防止早期 CFA 诱导的痛觉过敏。随后涉及神经元中 pPKCγ表达和激活的星形胶质细胞中 pERK2 和 IL-1β诱导的 NMDAR 非依赖性信号转导,有助于在 CFA 后 96 小时出现 NMDAR 非依赖性炎症性疼痛行为。有效减少炎症性疼痛的发生和维持需要针对这些研究中揭示的神经元-星形胶质细胞-细胞因子相互作用进行靶向治疗。