State Key Laboratory of Oral Diseases, National Clinical Research Center for Oral Diseases, West China Hospital of Stomatology, Sichuan University, Chengdu, China.
West China School of Stomatology Sichuan University, Chengdu, China.
Int J Oral Sci. 2019 Sep 10;11(3):24. doi: 10.1038/s41368-019-0055-0.
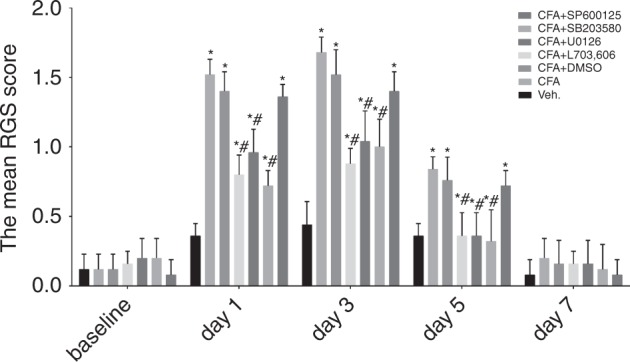
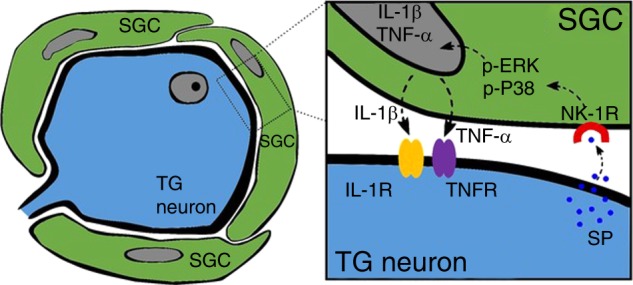
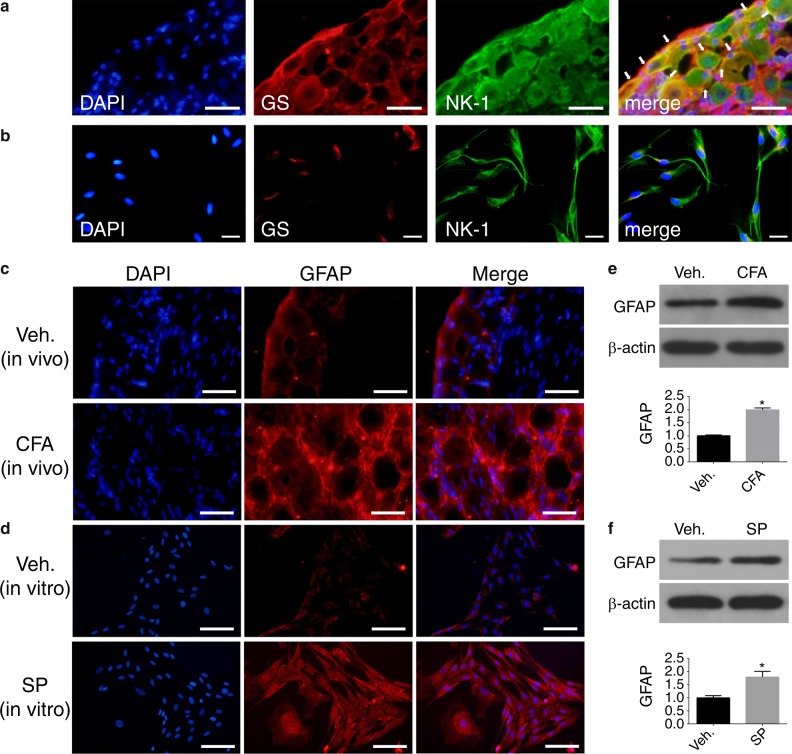
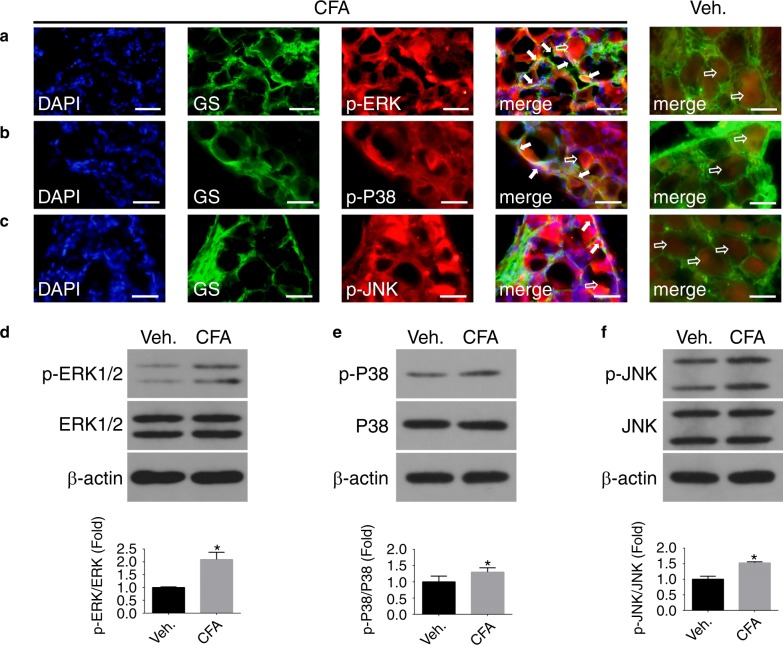
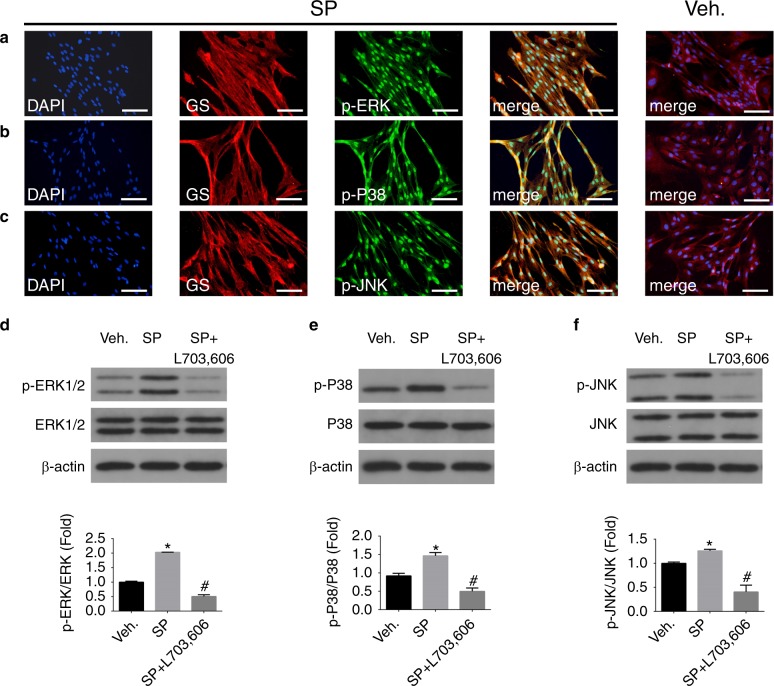
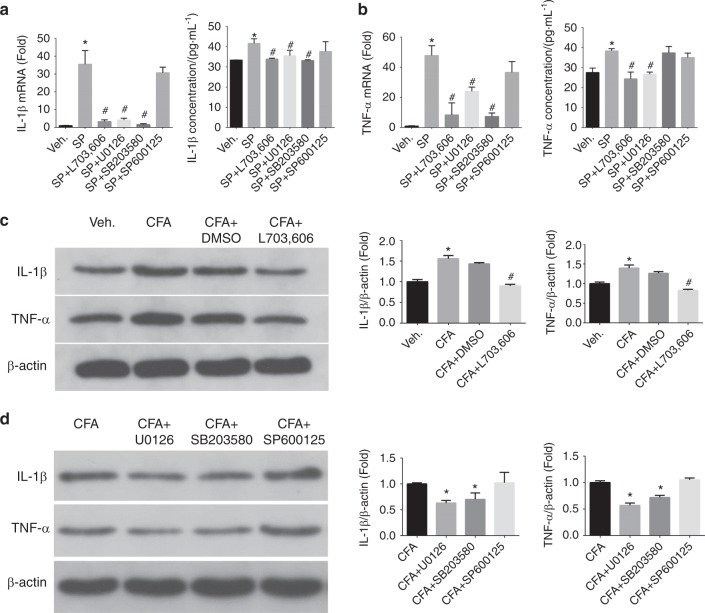
Inflammatory orofacial pain, in which substance P (SP) plays an important role, is closely related to the cross-talk between trigeminal ganglion (TG) neurons and satellite glial cells (SGCs). SGC activation is emerging as the key mechanism underlying inflammatory pain through different signalling mechanisms, including glial fibrillary acidic protein (GFAP) activation, phosphorylation of mitogen-activated protein kinase (MAPK) signalling pathways, and cytokine upregulation. However, in the TG, the mechanism underlying SP-mediated orofacial pain generated by SGCs is largely unknown. In this study, we investigated whether SP is involved in inflammatory orofacial pain by upregulating interleukin (IL)-1β and tumour necrosis factor (TNF)-α from SGCs, and we explored whether MAPK signalling pathways mediate the pain process. In the present study, complete Freund's adjuvant (CFA) was injected into the whisker pad of rats to induce an inflammatory model in vivo. SP was administered to SGC cultures in vitro to confirm the effect of SP. Facial expression analysis showed that pre-injection of L703,606 (an NK-1 receptor antagonist), U0126 (an inhibitor of MAPK/extracellular signal-regulated kinase [ERK] kinase [MEK] 1/2), and SB203580 (an inhibitor of P38) into the TG to induce targeted prevention of the activation of the NK-1 receptor and the phosphorylation of MAPKs significantly suppressed CFA-induced inflammatory allodynia. In addition, SP promoted SGC activation, which was proven by increased GFAP, p-MAPKs, IL-1β and TNF-α in SGCs under inflammatory conditions. Moreover, the increase in IL-1β and TNF-α was suppressed by L703, 606, U0126 and SB203580 in vivo and in vitro. These present findings suggested that SP, released from TG neurons, activated SGCs through the ERK1/2 and P38 pathways and promoted the production of IL-1β and TNF-α from SGCs, contributing to inflammatory orofacial pain associated with peripheral sensitization.
炎性面痛中,P 物质(SP)起着重要作用,其与三叉神经节(TG)神经元和卫星神经胶质细胞(SGC)之间的串扰密切相关。SGC 的激活被认为是炎症性疼痛的关键机制,其通过不同的信号机制,包括胶质纤维酸性蛋白(GFAP)的激活、丝裂原活化蛋白激酶(MAPK)信号通路的磷酸化和细胞因子的上调。然而,在 TG 中,SGC 产生的 SP 介导的炎性面痛的机制在很大程度上是未知的。在这项研究中,我们研究了 SP 是否通过上调 SGC 中的白细胞介素(IL)-1β和肿瘤坏死因子(TNF)-α来参与炎性面痛,并探讨了 MAPK 信号通路是否介导了疼痛过程。在本研究中,向大鼠触须垫注射完全弗氏佐剂(CFA)以在体内诱导炎症模型。在体外给予 SGC 培养物 SP,以确认 SP 的作用。面部表情分析表明,预先向 TG 注射 L703,606(NK-1 受体拮抗剂)、U0126(MAPK/细胞外信号调节激酶[ERK]激酶[MEK]1/2 抑制剂)和 SB203580(P38 抑制剂),以诱导 NK-1 受体和 MAPKs 磷酸化的靶向预防,显著抑制了 CFA 诱导的炎症性痛觉过敏。此外,SP 促进了 SGC 的激活,这通过在炎症条件下 SGC 中 GFAP、p-MAPKs、IL-1β和 TNF-α的增加得到证实。此外,体内和体外 L703,606、U0126 和 SB203580 抑制了 IL-1β 和 TNF-α 的增加。这些发现表明,SP 从 TG 神经元释放,通过 ERK1/2 和 P38 途径激活 SGC,并促进 SGC 产生 IL-1β 和 TNF-α,导致与外周敏化相关的炎性面痛。