Wellcome Trust Sanger Institute, Hinxton, CB10 1HH, UK.
Bioinformatics. 2010 Feb 15;26(4):565-7. doi: 10.1093/bioinformatics/btp693. Epub 2009 Dec 18.
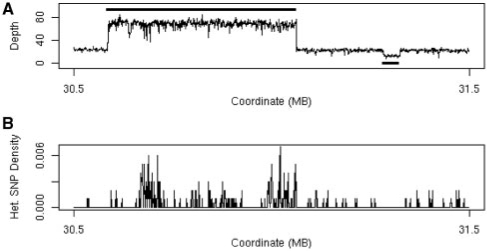
We have developed an algorithm to detect copy number variants (CNVs) in homozygous organisms, such as inbred laboratory strains of mice, from short read sequence data. Our novel approach exploits the fact that inbred mice are homozygous at virtually every position in the genome to detect CNVs using a hidden Markov model (HMM). This HMM uses both the density of sequence reads mapped to the genome, and the rate of apparent heterozygous single nucleotide polymorphisms, to determine genomic copy number. We tested our algorithm on short read sequence data generated from re-sequencing chromosome 17 of the mouse strains A/J and CAST/EiJ with the Illumina platform. In total, we identified 118 copy number variants (43 for A/J and 75 for CAST/EiJ). We investigated the performance of our algorithm through comparison to CNVs previously identified by array-comparative genomic hybridization (array CGH). We performed quantitative-PCR validation on a subset of the calls that differed from the array CGH data sets.
我们开发了一种算法,用于从短读序列数据中检测纯合生物体(如近交系实验室小鼠)中的拷贝数变异(CNV)。我们的新方法利用近交系小鼠在基因组的几乎每个位置都是纯合子的事实,使用隐马尔可夫模型(HMM)来检测 CNV。该 HMM 同时使用映射到基因组的序列读取密度和明显杂合单核苷酸多态性的速率来确定基因组拷贝数。我们使用 Illumina 平台对来自 A/J 和 CAST/EiJ 小鼠品系的第 17 号染色体进行重测序的短读序列数据测试了我们的算法。总共鉴定出 118 个拷贝数变异(A/J 为 43 个,CAST/EiJ 为 75 个)。我们通过与先前通过阵列比较基因组杂交(array CGH)鉴定的 CNV 进行比较来评估我们算法的性能。我们对与 array CGH 数据集不同的部分调用进行了定量-PCR 验证。