Cahan Patrick, Godfrey Laura E, Eis Peggy S, Richmond Todd A, Selzer Rebecca R, Brent Michael, McLeod Howard L, Ley Timothy J, Graubert Timothy A
Department of Internal Medicine and Department of Genetics, Division of Oncology, Stem Cell Biology Section, Washington University, St Louis, MO, USA.
Nucleic Acids Res. 2008 Apr;36(7):e41. doi: 10.1093/nar/gkn110. Epub 2008 Mar 11.
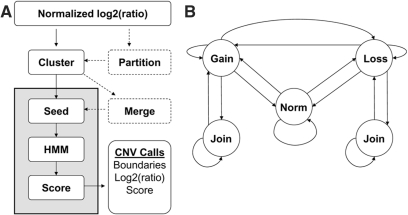
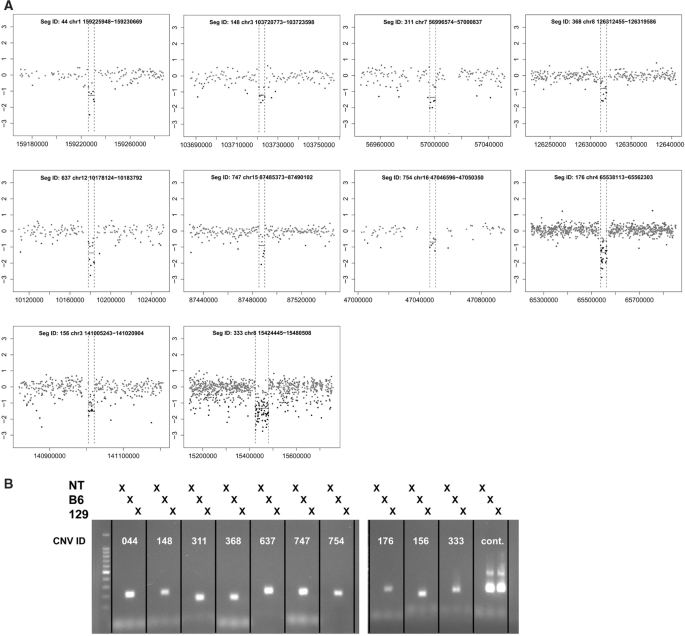
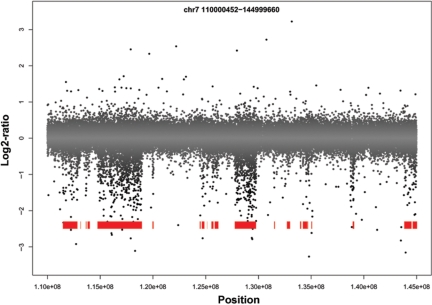
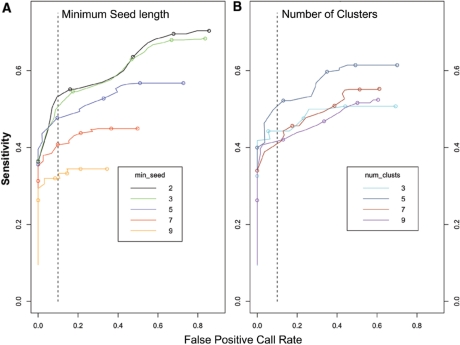
Copy number variants (CNVs) are currently defined as genomic sequences that are polymorphic in copy number and range in length from 1000 to several million base pairs. Among current array-based CNV detection platforms, long-oligonucleotide arrays promise the highest resolution. However, the performance of currently available analytical tools suffers when applied to these data because of the lower signal:noise ratio inherent in oligonucleotide-based hybridization assays. We have developed wuHMM, an algorithm for mapping CNVs from array comparative genomic hybridization (aCGH) platforms comprised of 385 000 to more than 3 million probes. wuHMM is unique in that it can utilize sequence divergence information to reduce the false positive rate (FPR). We apply wuHMM to 385K-aCGH, 2.1M-aCGH and 3.1M-aCGH experiments comparing the 129X1/SvJ and C57BL/6J inbred mouse genomes. We assess wuHMM's performance on the 385K platform by comparison to the higher resolution platforms and we independently validate 10 CNVs. The method requires no training data and is robust with respect to changes in algorithm parameters. At a FPR of <10%, the algorithm can detect CNVs with five probes on the 385K platform and three on the 2.1M and 3.1M platforms, resulting in effective resolutions of 24 kb, 2-5 kb and 1 kb, respectively.
拷贝数变异(CNV)目前被定义为拷贝数呈多态性且长度范围从1000个碱基对到数百万个碱基对的基因组序列。在当前基于阵列的CNV检测平台中,长寡核苷酸阵列有望实现最高分辨率。然而,由于基于寡核苷酸的杂交检测中固有的较低信噪比,当前可用的分析工具在应用于这些数据时性能不佳。我们开发了wuHMM,这是一种用于从由385,000个到超过300万个探针组成的阵列比较基因组杂交(aCGH)平台映射CNV的算法。wuHMM的独特之处在于它可以利用序列差异信息来降低假阳性率(FPR)。我们将wuHMM应用于比较129X1/SvJ和C57BL/6J近交小鼠基因组的385K-aCGH、2.1M-aCGH和3.1M-aCGH实验。我们通过与更高分辨率平台比较来评估wuHMM在385K平台上的性能,并独立验证了10个CNV。该方法不需要训练数据,并且在算法参数变化方面具有鲁棒性。在FPR<10%时,该算法在385K平台上可以用五个探针检测CNV,在2.1M和3.1M平台上可以用三个探针检测CNV,有效分辨率分别为24 kb、2 - 5 kb和1 kb。