Imprialou Martha, Kahles André, Steffen Joshua G, Osborne Edward J, Gan Xiangchao, Lempe Janne, Bhomra Amarjit, Belfield Eric, Visscher Anne, Greenhalgh Robert, Harberd Nicholas P, Goram Richard, Hein Jotun, Robert-Seilaniantz Alexandre, Jones Jonathan, Stegle Oliver, Kover Paula, Tsiantis Miltos, Nordborg Magnus, Rätsch Gunnar, Clark Richard M, Mott Richard
Wellcome Trust Centre for Human Genetics, University of Oxford, OX3 7BN, United Kingdom.
Department of Statistics, University of Oxford, OX1 3TG, United Kingdom.
Genetics. 2017 Apr;205(4):1425-1441. doi: 10.1534/genetics.116.192823. Epub 2017 Feb 7.
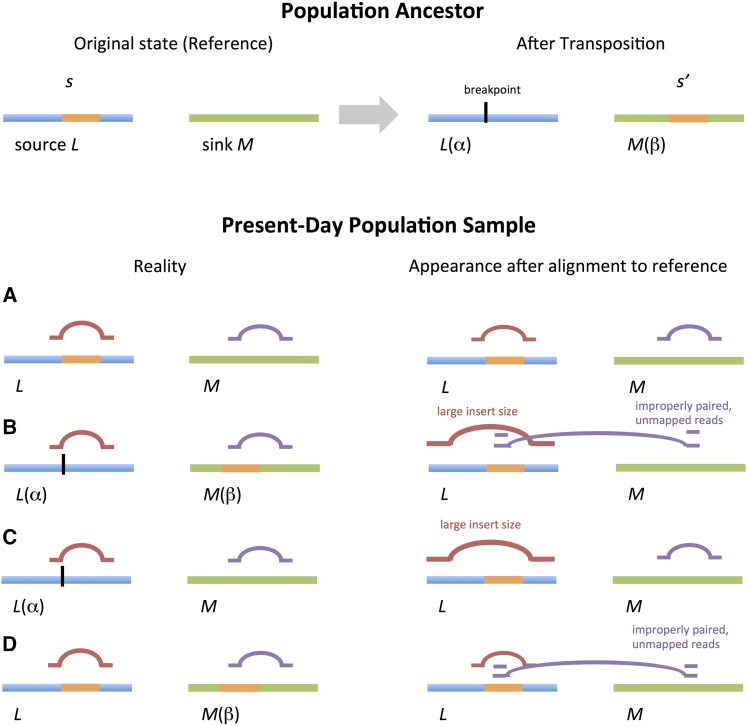
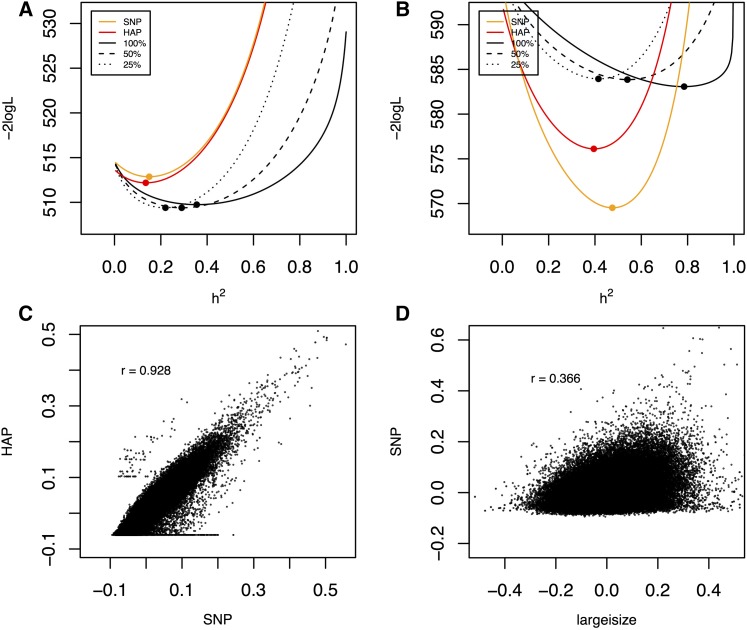
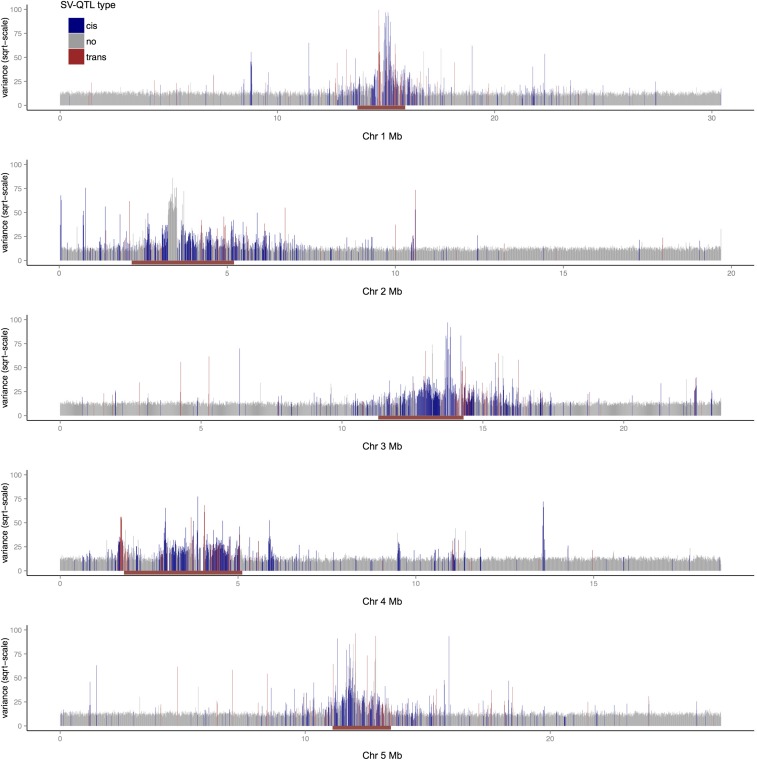
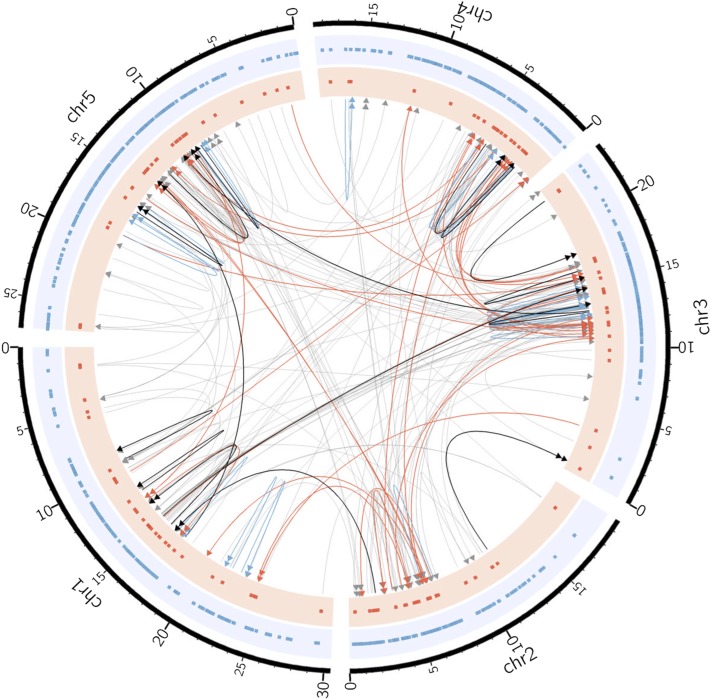
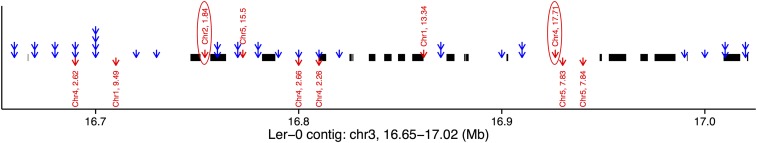
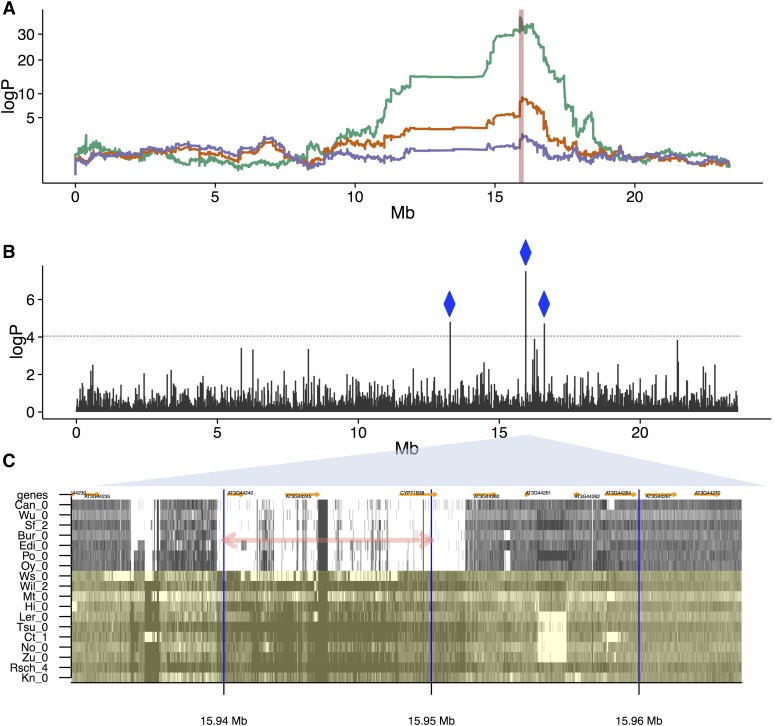
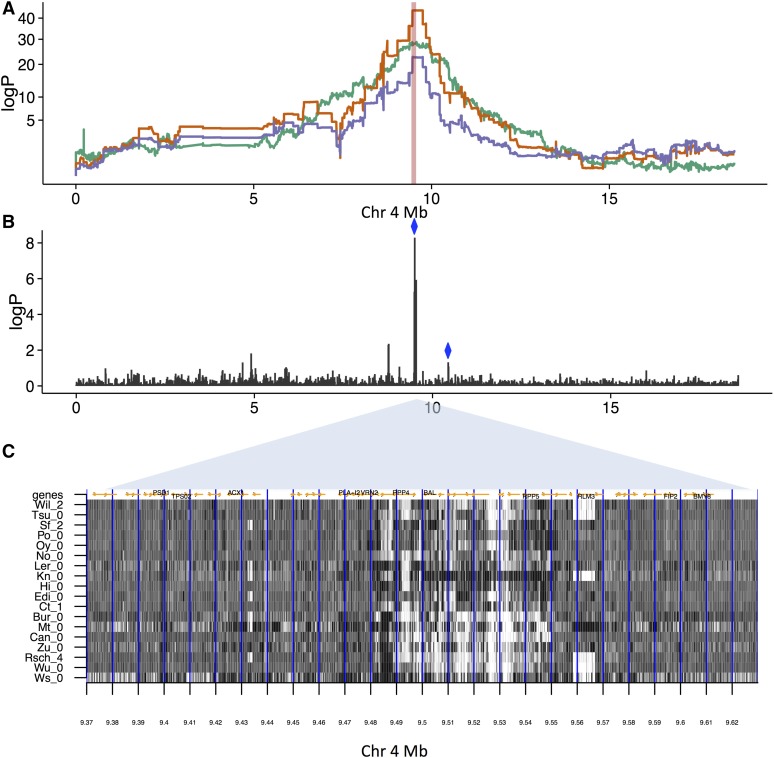
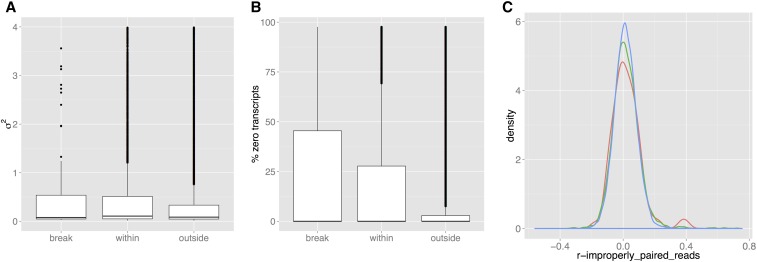
To understand the population genetics of structural variants and their effects on phenotypes, we developed an approach to mapping structural variants that segregate in a population sequenced at low coverage. We avoid calling structural variants directly. Instead, the evidence for a potential structural variant at a locus is indicated by variation in the counts of short-reads that map anomalously to that locus. These structural variant traits are treated as quantitative traits and mapped genetically, analogously to a gene expression study. Association between a structural variant trait at one locus, and genotypes at a distant locus indicate the origin and target of a transposition. Using ultra-low-coverage (0.3×) population sequence data from 488 recombinant inbred genomes, we identified 6502 segregating structural variants. Remarkably, 25% of these were transpositions. While many structural variants cannot be delineated precisely, we validated 83% of 44 predicted transposition breakpoints by polymerase chain reaction. We show that specific structural variants may be causative for quantitative trait loci for germination and resistance to infection by the fungus , isolate Nc14. Further we show that the phenotypic heritability attributable to read-mapping anomalies differs from, and, in the case of time to germination and bolting, exceeds that due to standard genetic variation. Genes within structural variants are also more likely to be silenced or dysregulated. This approach complements the prevalent strategy of structural variant discovery in fewer individuals sequenced at high coverage. It is generally applicable to large populations sequenced at low-coverage, and is particularly suited to mapping transpositions.
为了理解结构变异的群体遗传学及其对表型的影响,我们开发了一种在低覆盖度测序的群体中定位分离的结构变异的方法。我们避免直接调用结构变异。相反,某个位点潜在结构变异的证据由异常映射到该位点的短读段计数的变化来指示。这些结构变异性状被视为数量性状并进行遗传定位,类似于基因表达研究。一个位点的结构变异性状与远处位点的基因型之间的关联表明了转座的起源和靶点。利用来自488个重组近交基因组的超低覆盖度(0.3×)群体序列数据,我们鉴定出6502个分离的结构变异。值得注意的是,其中25%是转座。虽然许多结构变异无法精确界定,但我们通过聚合酶链反应验证了44个预测的转座断点中的83%。我们表明,特定的结构变异可能是发芽和对真菌Nc14感染抗性的数量性状位点的原因。此外,我们表明,归因于读段映射异常的表型遗传力与标准遗传变异不同,并且在发芽和抽薹时间方面超过了标准遗传变异。结构变异内的基因也更有可能被沉默或失调。这种方法补充了在少数高覆盖度测序个体中发现结构变异的普遍策略。它通常适用于低覆盖度测序的大群体,特别适合于转座的定位。