Le Ly, Lee Eric, Schulten Klaus, Truong Thanh N
Department of Chemistry, University of Utah; Beckman Institute for Advanced Science and Technology and Professor of Chemistry, University of Utah, & Director of ICST, Vietnam.
PLoS Curr. 2009 Aug 27;1:RRN1015. doi: 10.1371/currents.RRN1015.
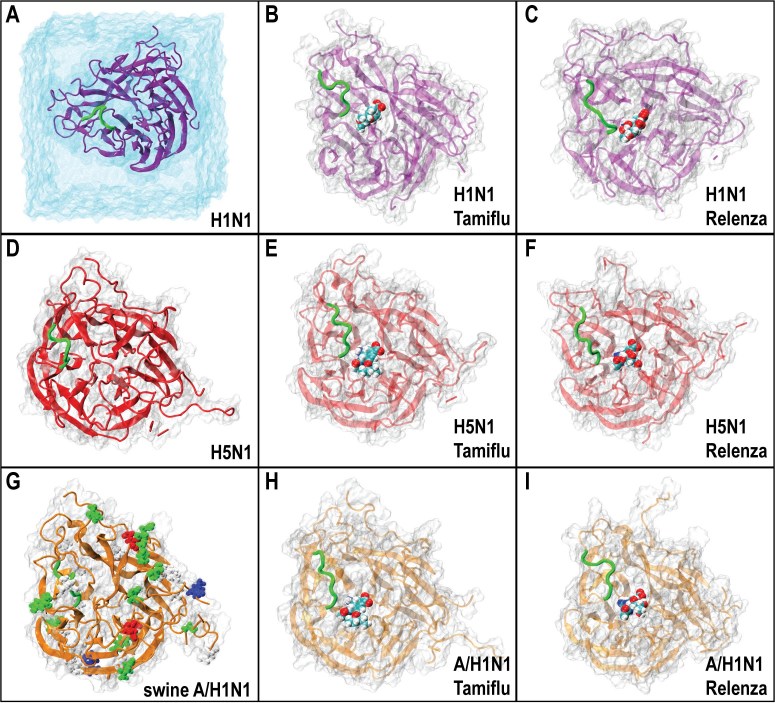
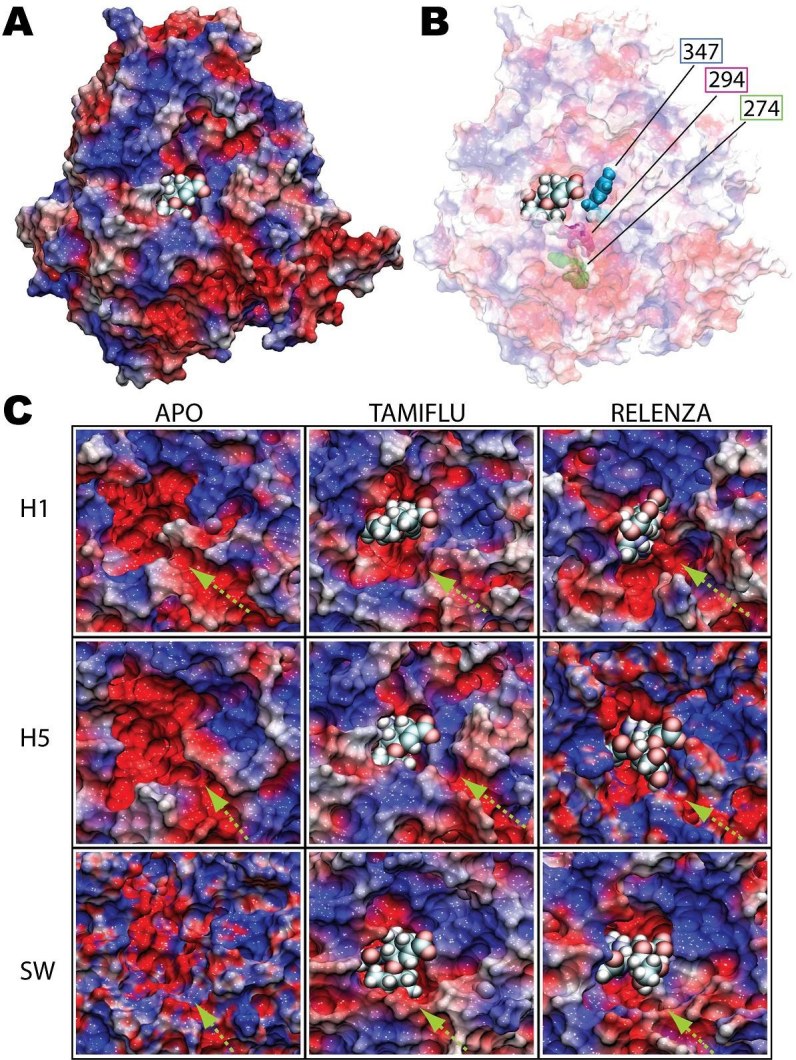
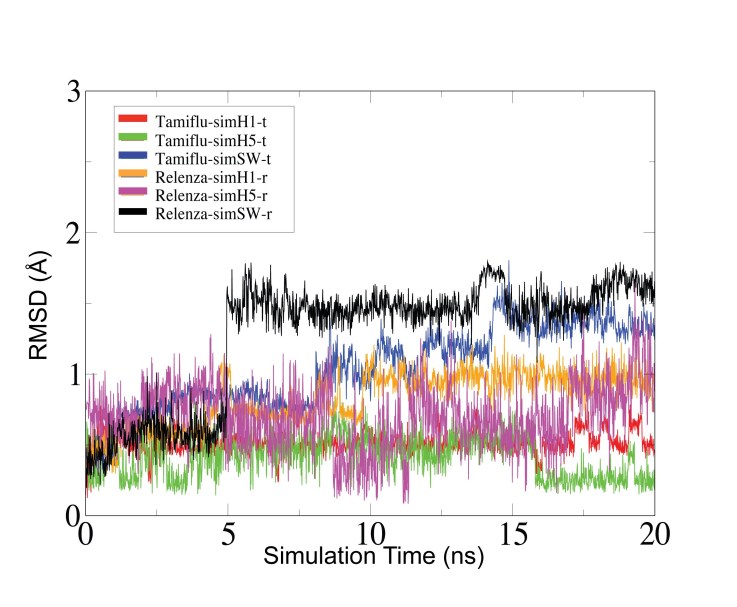
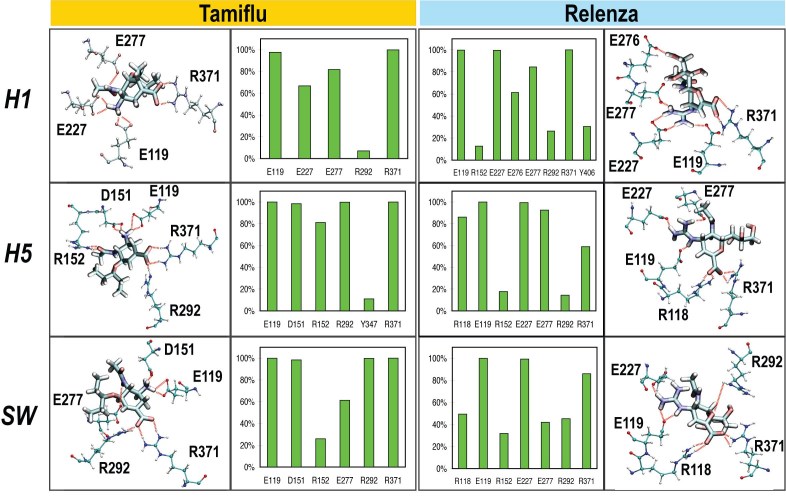
A molecular model of the swine influenza A/H1N1 ( also called H1N1pdm) type-I neuraminidase was built using the pathogenic avian H5N1 type-I neuraminidase as a basis, due to the higher sequence identity between A/H1N1 and H5N1 (91.47%) compared to Spanish H1N1 (88.37%) neuraminidase. All-atom molecular dynamics (MD) simulations of all three neuraminidases were performed, either as apo-structures or with commercial antiviral drugs Tamiflu or Relenza separately bound; the simulations allowed for the identification of both conserved and unique drug-protein interactions across all three proteins. Specifically, conserved networks of hydrogen bonds stabilizing the drugs in the sialic acid binding site of the simulated neuraminidases are analyzed, providing insight into how disruption due to mutations may lead to increased drug resistance. In addition, a possible mechanism through which the residue 294 mutation acquires drug resistance is proposed by mapping the mutation site onto an electrostatic pathway which may play a role in controlling drug access to the binding pocket of neuraminidase, establishing a starting point for further investigations of neuraminidase drug resistance.
由于甲型H1N1流感病毒(也称为H1N1pdm)与致病性禽流感H5N1病毒的I型神经氨酸酶之间的序列同一性(91.47%)高于西班牙H1N1病毒(88.37%)的神经氨酸酶,因此以致病性禽流感H5N1病毒的I型神经氨酸酶为基础构建了甲型H1N1流感病毒的分子模型。对所有三种神经氨酸酶进行了全原子分子动力学(MD)模拟,模拟形式为无配体结构,或分别与商业抗病毒药物达菲或瑞乐沙结合;这些模拟有助于识别所有三种蛋白质中保守和独特的药物-蛋白质相互作用。具体而言,分析了模拟神经氨酸酶唾液酸结合位点中稳定药物的氢键保守网络,从而深入了解突变引起的破坏如何可能导致耐药性增加。此外,通过将突变位点映射到可能在控制药物进入神经氨酸酶结合口袋中起作用的静电通路上,提出了294位残基突变获得耐药性的可能机制,为进一步研究神经氨酸酶耐药性奠定了基础。