Tanner David E, Phillips James C, Schulten Klaus
Center for Biophysics and Computational Biology, University of Illinois at Urbana-Champaign ; Beckman Institute, University of Illinois at Urbana-Champaign.
J Chem Theory Comput. 2012 Jul 10;8(7):2521-2530. doi: 10.1021/ct3003089. Epub 2012 Jun 15.
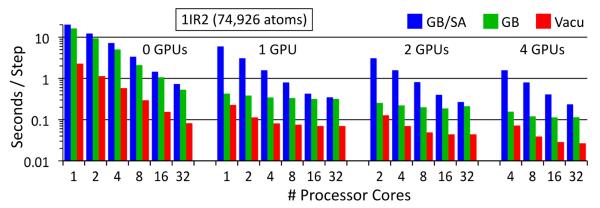
Molecular dynamics methodologies comprise a vital research tool for structural biology. Molecular dynamics has benefited from technological advances in computing, such as multi-core CPUs and graphics processing units (GPUs), but harnessing the full power of hybrid GPU/CPU computers remains difficult. The generalized Born/solvent-accessible surface area implicit solvent model (GB/SA) stands to benefit from hybrid GPU/CPU computers, employing the GPU for the GB calculation and the CPU for the SA calculation. Here, we explore the computational challenges facing GB/SA calculations on hybrid GPU/CPU computers and demonstrate how NAMD, a parallel molecular dynamics program, is able to efficiently utilize GPUs and CPUs simultaneously for fast GB/SA simulations. The hybrid computation principles demonstrated here are generally applicable to parallel applications employing hybrid GPU/CPU calculations.
分子动力学方法是结构生物学的重要研究工具。分子动力学受益于计算技术的进步,如多核CPU和图形处理单元(GPU),但充分利用混合GPU/CPU计算机的全部性能仍然困难。广义玻恩/溶剂可及表面积隐式溶剂模型(GB/SA)有望从混合GPU/CPU计算机中受益,利用GPU进行GB计算,利用CPU进行SA计算。在这里,我们探讨了在混合GPU/CPU计算机上进行GB/SA计算所面临的计算挑战,并展示了并行分子动力学程序NAMD如何能够同时高效地利用GPU和CPU进行快速GB/SA模拟。这里展示的混合计算原理通常适用于采用混合GPU/CPU计算的并行应用程序。