Center for Human Genome Variation, Duke Institute for Genome Sciences and Policy, Duke University, Durham, North Carolina, USA.
PLoS Genet. 2009 Dec;5(12):e1000791. doi: 10.1371/journal.pgen.1000791. Epub 2009 Dec 24.
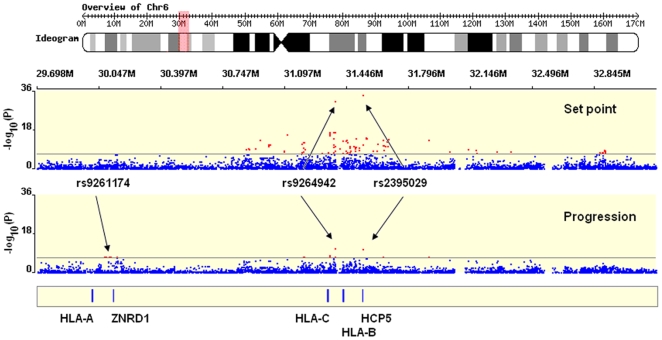
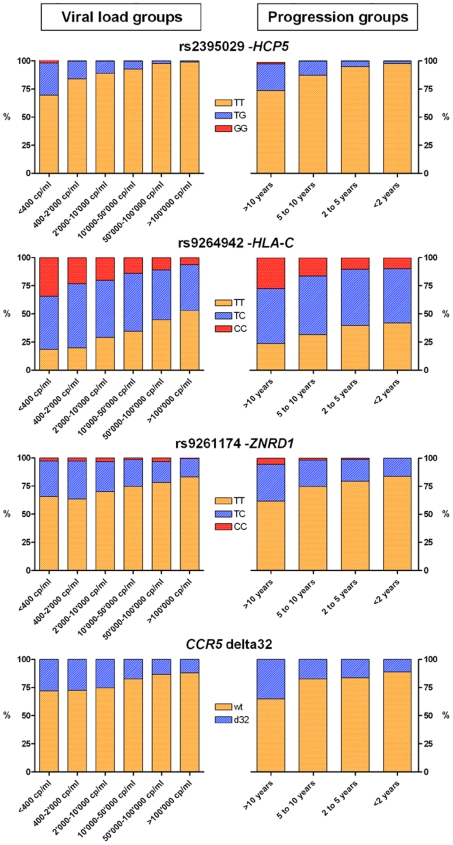
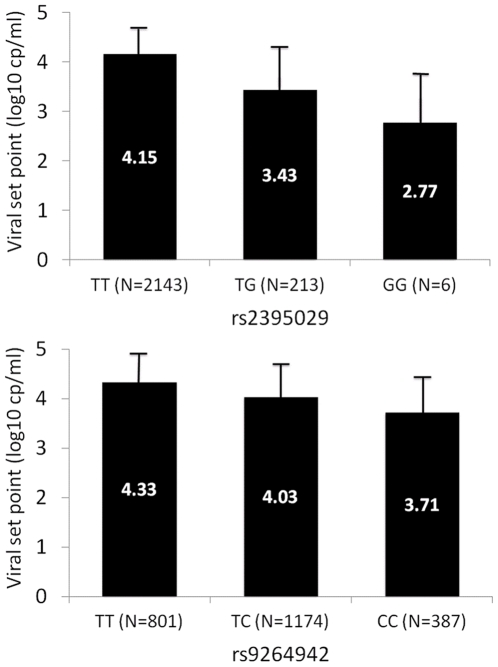
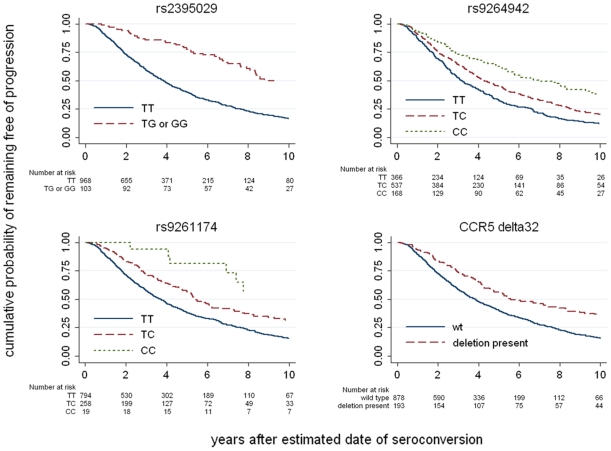
To extend the understanding of host genetic determinants of HIV-1 control, we performed a genome-wide association study in a cohort of 2,554 infected Caucasian subjects. The study was powered to detect common genetic variants explaining down to 1.3% of the variability in viral load at set point. We provide overwhelming confirmation of three associations previously reported in a genome-wide study and show further independent effects of both common and rare variants in the Major Histocompatibility Complex region (MHC). We also examined the polymorphisms reported in previous candidate gene studies and fail to support a role for any variant outside of the MHC or the chemokine receptor cluster on chromosome 3. In addition, we evaluated functional variants, copy-number polymorphisms, epistatic interactions, and biological pathways. This study thus represents a comprehensive assessment of common human genetic variation in HIV-1 control in Caucasians.
为了更深入地了解宿主遗传因素对 HIV-1 控制的影响,我们对 2554 名感染的白种人受试者进行了全基因组关联研究。该研究的目的是检测常见的遗传变异,以解释在固定点时病毒载量可降低 1.3%的变异。我们对全基因组研究中以前报道的三个关联进行了压倒性的确认,并进一步显示了主要组织相容性复合体(MHC)区域中常见和罕见变异的独立影响。我们还检查了以前候选基因研究中报道的多态性,但没有支持 MHC 以外或染色体 3 上趋化因子受体簇的任何变异的作用。此外,我们还评估了功能变体、拷贝数多态性、上位性相互作用和生物学途径。因此,这项研究代表了对白人中 HIV-1 控制的常见人类遗传变异的全面评估。