Department of Biochemistry, Universidad Autónoma de Madrid-Instituto de Investigaciones Biomédicas CSIC, Madrid, Spain.
Nucleic Acids Res. 2010 Apr;38(7):2332-45. doi: 10.1093/nar/gkp1205. Epub 2010 Jan 8.
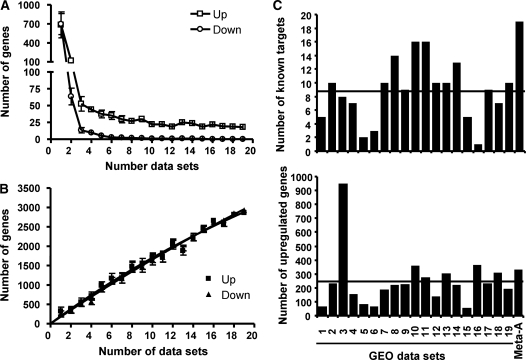
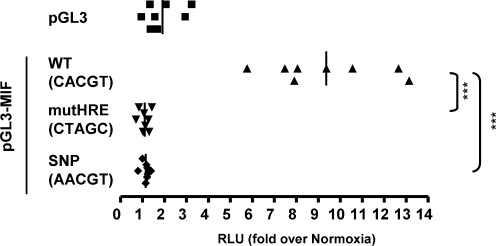
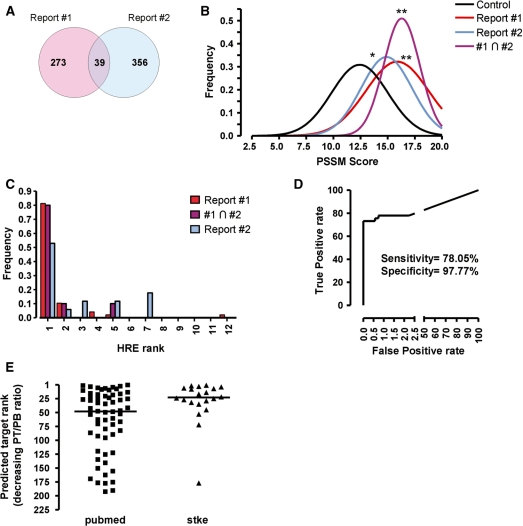
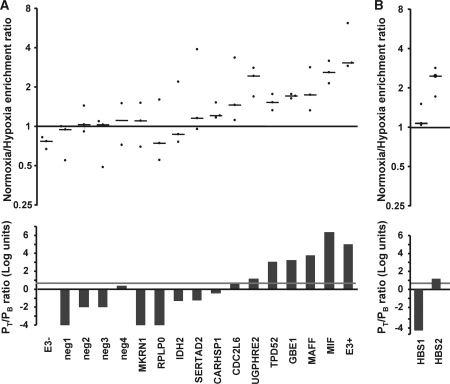
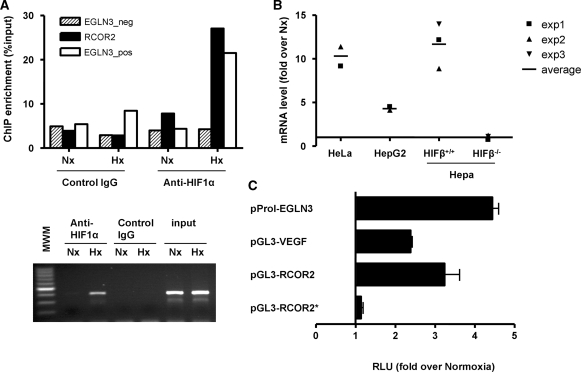
The transcriptional response driven by Hypoxia-inducible factor (HIF) is central to the adaptation to oxygen restriction. Hence, the complete identification of HIF targets is essential for understanding the cellular responses to hypoxia. Herein we describe a computational strategy based on the combination of phylogenetic footprinting and transcription profiling meta-analysis for the identification of HIF-target genes. Comparison of the resulting candidates with published HIF1a genome-wide chromatin immunoprecipitation indicates a high sensitivity (78%) and specificity (97.8%). To validate our strategy, we performed HIF1a chromatin immunoprecipitation on a set of putative targets. Our results confirm the robustness of the computational strategy in predicting HIF-binding sites and reveal several novel HIF targets, including RE1-silencing transcription factor co-repressor (RCOR2). In addition, mapping of described polymorphisms to the predicted HIF-binding sites identified several single-nucleotide polymorphisms (SNPs) that could alter HIF binding. As a proof of principle, we demonstrate that SNP rs17004038, mapping to a functional hypoxia response element in the macrophage migration inhibitory factor (MIF) locus, prevents induction of this gene by hypoxia. Altogether, our results show that the proposed strategy is a powerful tool for the identification of HIF direct targets that expands our knowledge of the cellular adaptation to hypoxia and provides cues on the inter-individual variation in this response.
缺氧诱导因子 (HIF) 驱动的转录反应是适应氧限制的核心。因此,完整识别 HIF 靶标对于理解细胞对缺氧的反应至关重要。在此,我们描述了一种基于系统发育足迹和转录谱荟萃分析相结合的计算策略,用于鉴定 HIF 靶基因。将得到的候选基因与已发表的 HIF1a 全基因组染色质免疫沉淀进行比较,表明该方法具有较高的灵敏度(78%)和特异性(97.8%)。为了验证我们的策略,我们对一组假定的靶基因进行了 HIF1a 染色质免疫沉淀。我们的结果证实了该计算策略在预测 HIF 结合位点方面的稳健性,并揭示了几个新的 HIF 靶基因,包括 RE1 沉默转录因子共抑制因子 (RCOR2)。此外,将描述的多态性映射到预测的 HIF 结合位点,确定了几个可能改变 HIF 结合的单核苷酸多态性 (SNP)。作为原理验证,我们证明了 SNP rs17004038 映射到巨噬细胞移动抑制因子 (MIF) 基因座中功能性缺氧反应元件,可阻止该基因在缺氧时的诱导。总之,我们的结果表明,所提出的策略是鉴定 HIF 直接靶标的有力工具,可扩展我们对细胞适应缺氧的认识,并为该反应中的个体间差异提供线索。