Department of Genetics and Biometry, Research Institute for Biology of Farm Animals, Wilhelm-Stahl Allee 2, D 18196 Dummerstorf, Germany.
BMC Bioinformatics. 2010 Jan 14;11:27. doi: 10.1186/1471-2105-11-27.
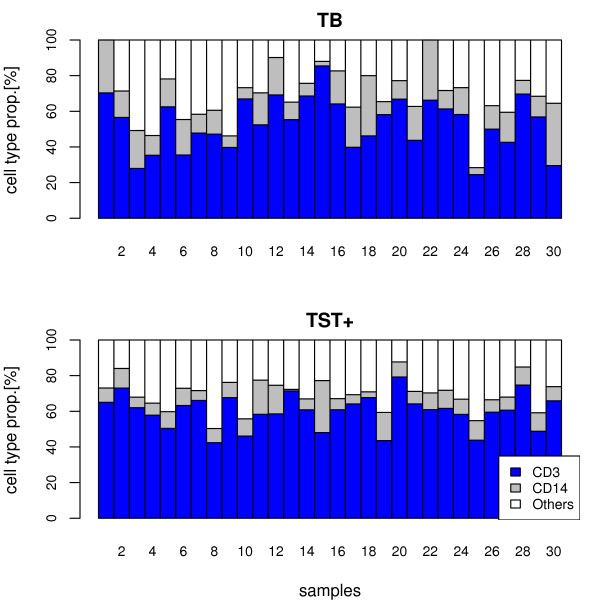
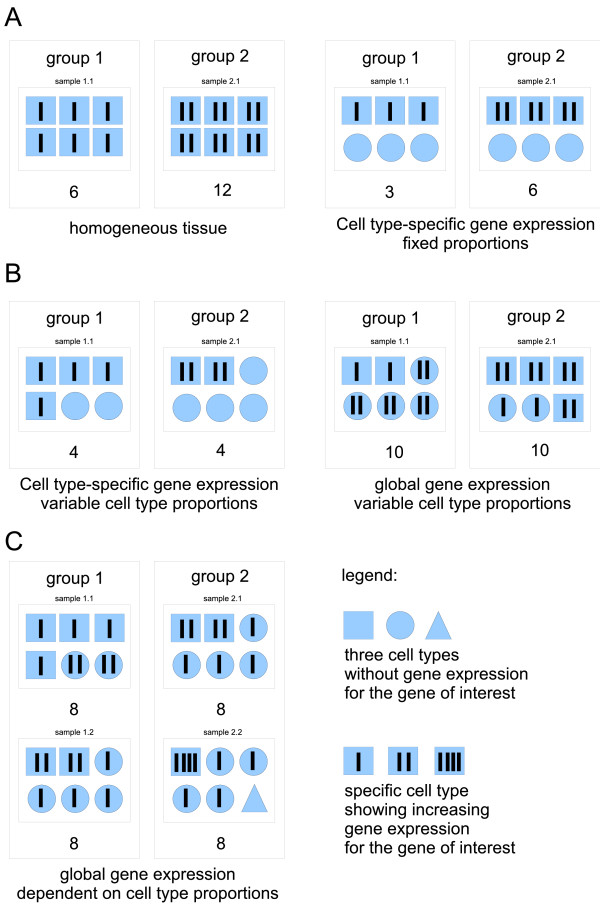
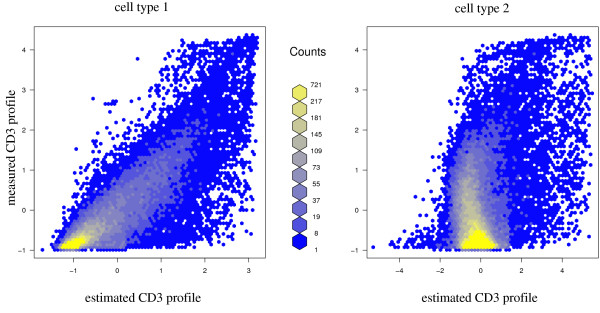
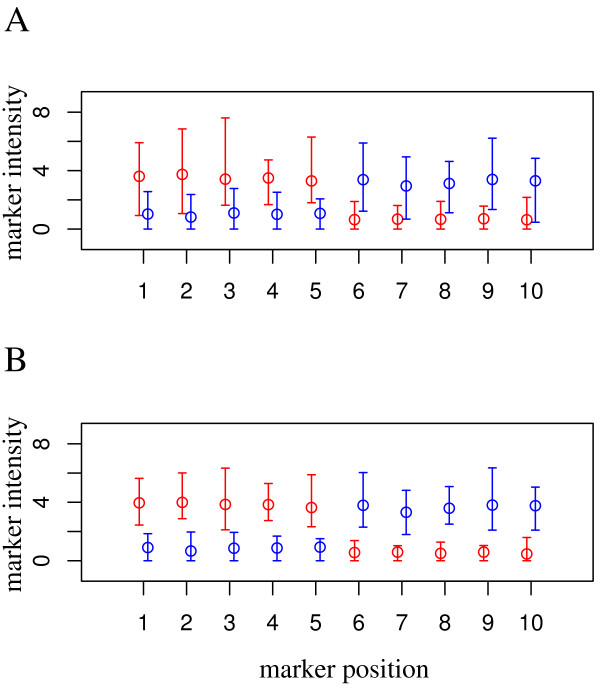
For heterogeneous tissues, such as blood, measurements of gene expression are confounded by relative proportions of cell types involved. Conclusions have to rely on estimation of gene expression signals for homogeneous cell populations, e.g. by applying micro-dissection, fluorescence activated cell sorting, or in-silico deconfounding. We studied feasibility and validity of a non-negative matrix decomposition algorithm using experimental gene expression data for blood and sorted cells from the same donor samples. Our objective was to optimize the algorithm regarding detection of differentially expressed genes and to enable its use for classification in the difficult scenario of reversely regulated genes. This would be of importance for the identification of candidate biomarkers in heterogeneous tissues.
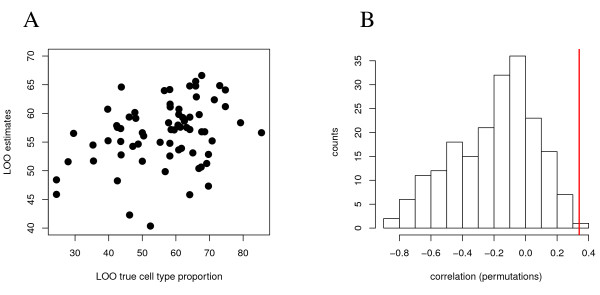
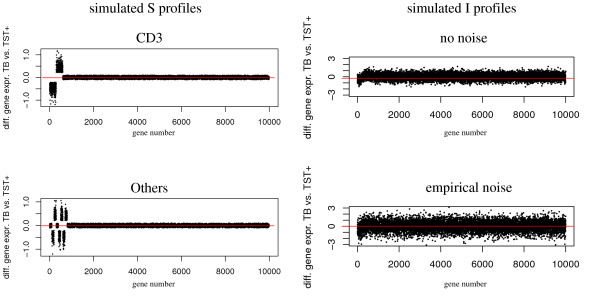
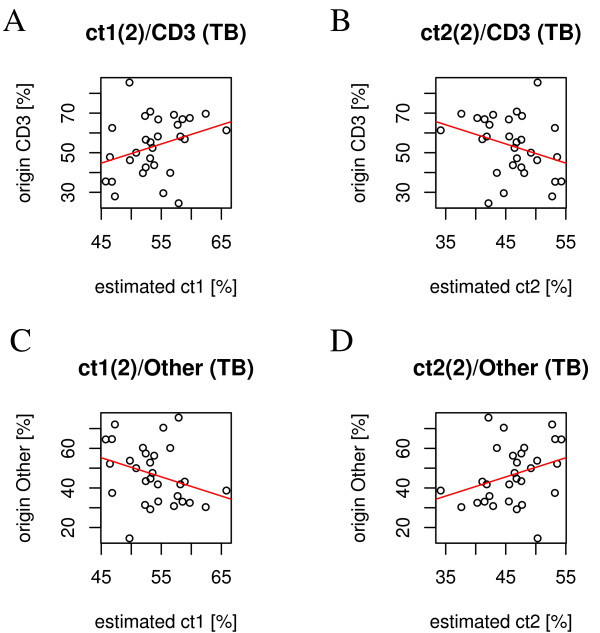
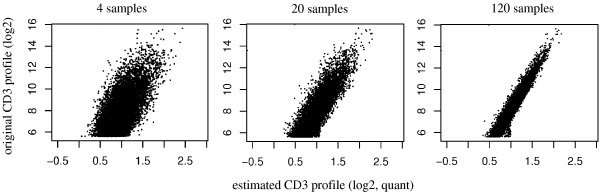
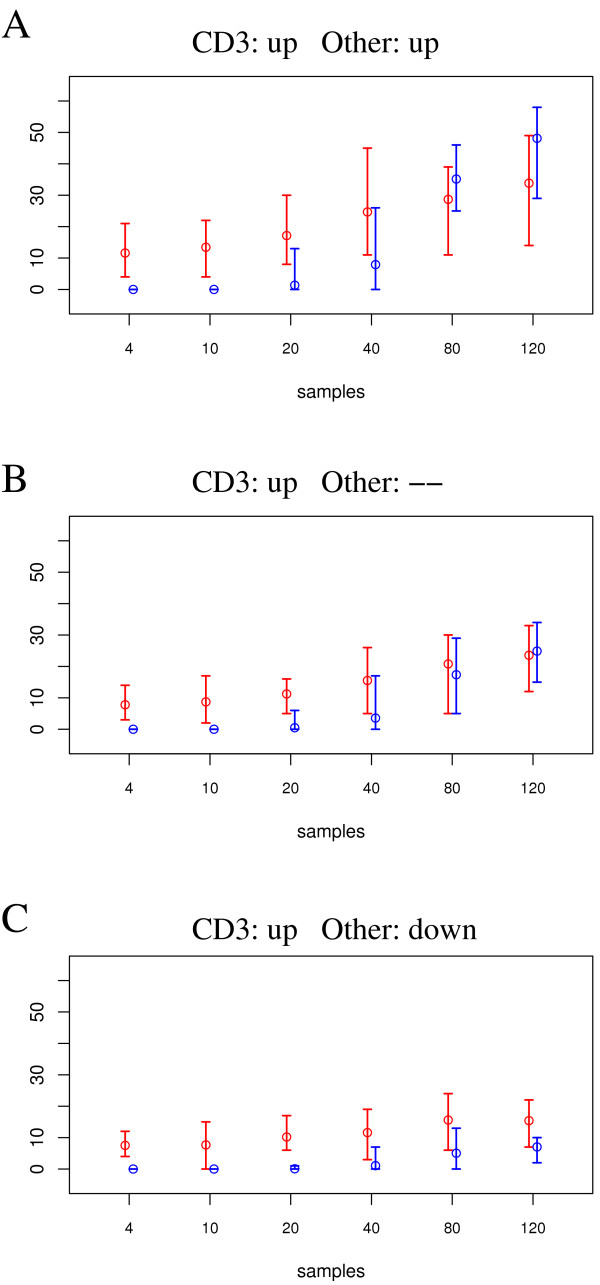
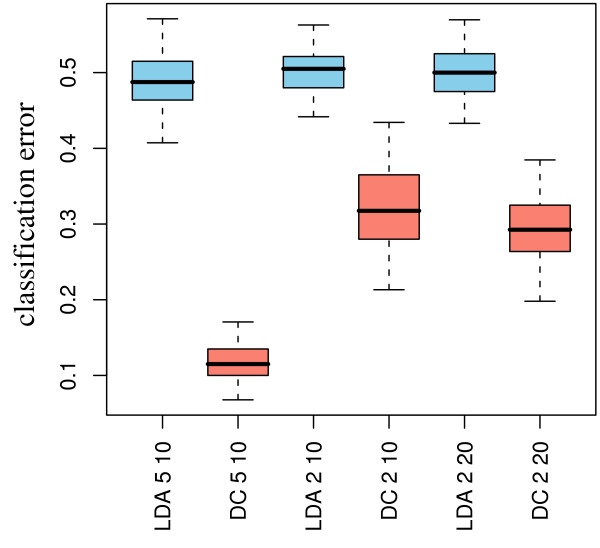
Experimental data and simulation studies involving noise parameters estimated from these data revealed that for valid detection of differential gene expression, quantile normalization and use of non-log data are optimal. We demonstrate the feasibility of predicting proportions of constituting cell types from gene expression data of single samples, as a prerequisite for a deconfounding-based classification approach.Classification cross-validation errors with and without using deconfounding results are reported as well as sample-size dependencies. Implementation of the algorithm, simulation and analysis scripts are available.
The deconfounding algorithm without decorrelation using quantile normalization on non-log data is proposed for biomarkers that are difficult to detect, and for cases where confounding by varying proportions of cell types is the suspected reason. In this case, a deconfounding ranking approach can be used as a powerful alternative to, or complement of, other statistical learning approaches to define candidate biomarkers for molecular diagnosis and prediction in biomedicine, in realistically noisy conditions and with moderate sample sizes.
对于异质组织,如血液,基因表达的测量受到涉及的细胞类型相对比例的影响。结论必须依赖于对同质细胞群体的基因表达信号的估计,例如通过应用显微切割、荧光激活细胞分选或计算机去混淆。我们使用来自同一供体样本的血液和分选细胞的实验基因表达数据研究了非负矩阵分解算法的可行性和有效性。我们的目的是优化该算法,以检测差异表达基因,并使其能够用于在反向调节基因的困难情况下进行分类。这对于在异质组织中识别候选生物标志物非常重要。
实验数据和涉及从这些数据估计的噪声参数的模拟研究表明,对于有效检测差异基因表达,分位数归一化和使用非对数数据是最优的。我们证明了从单个样本的基因表达数据预测构成细胞类型比例的可行性,这是去混淆分类方法的前提。报告了有无去混淆结果的分类交叉验证错误以及样本大小依赖性。该算法的实现、模拟和分析脚本都可用。
提出了一种使用分位数归一化和非对数数据的非相关去混淆算法,用于难以检测的生物标志物,以及怀疑细胞类型比例变化是混杂因素的情况。在这种情况下,去混淆排序方法可以作为其他统计学习方法的替代方法或补充方法,用于在合理噪声条件下和中等样本量下定义生物医学分子诊断和预测的候选生物标志物。