Department of Neurology, Children's Hospital Boston, Boston, MA 02115, USA.
Cell. 2010 Jan 8;140(1):74-87. doi: 10.1016/j.cell.2009.12.011.
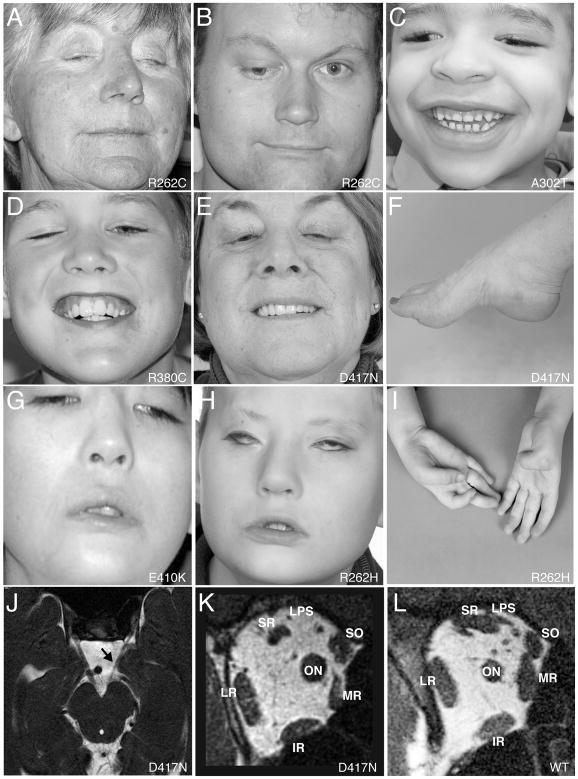
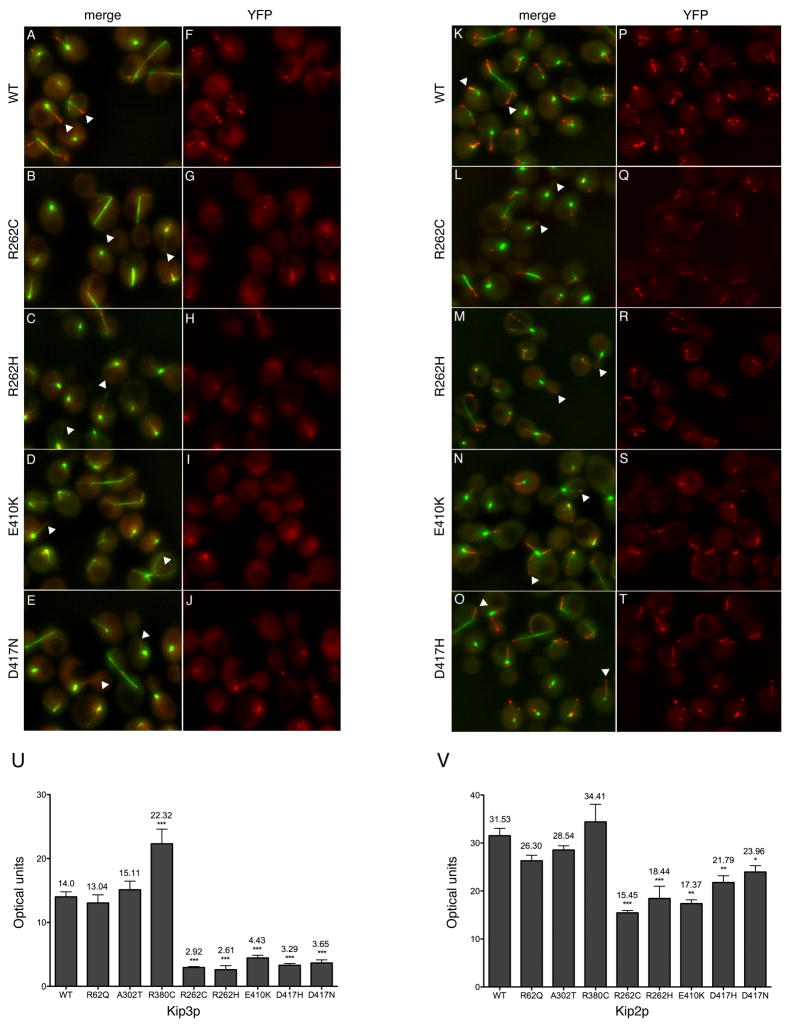
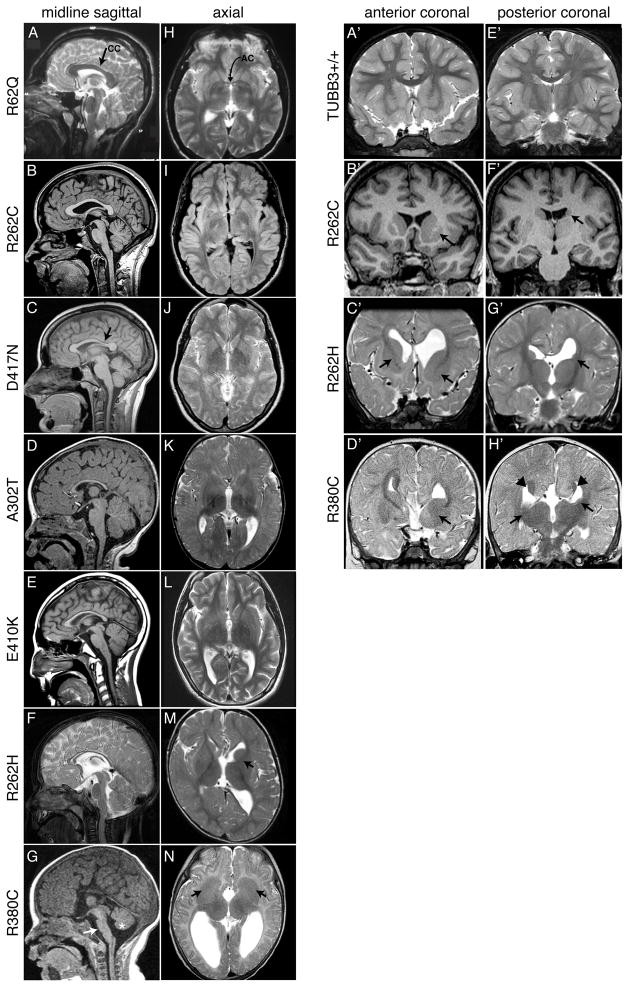
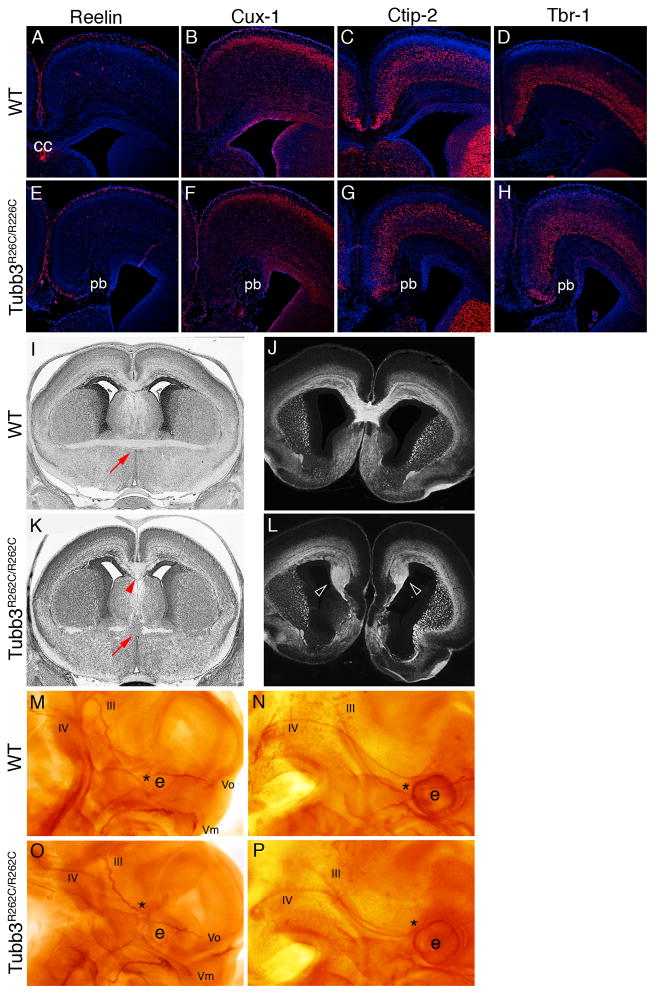
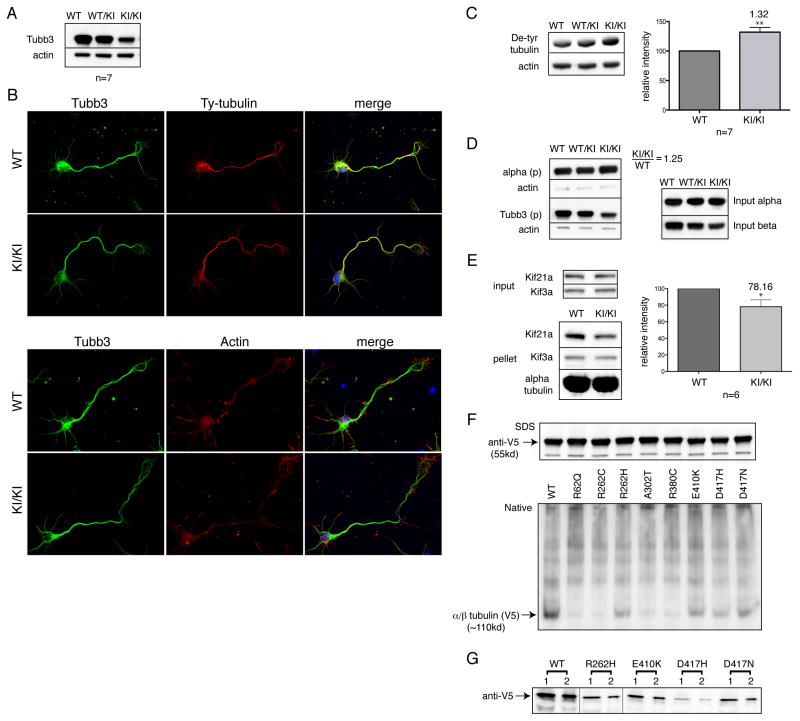
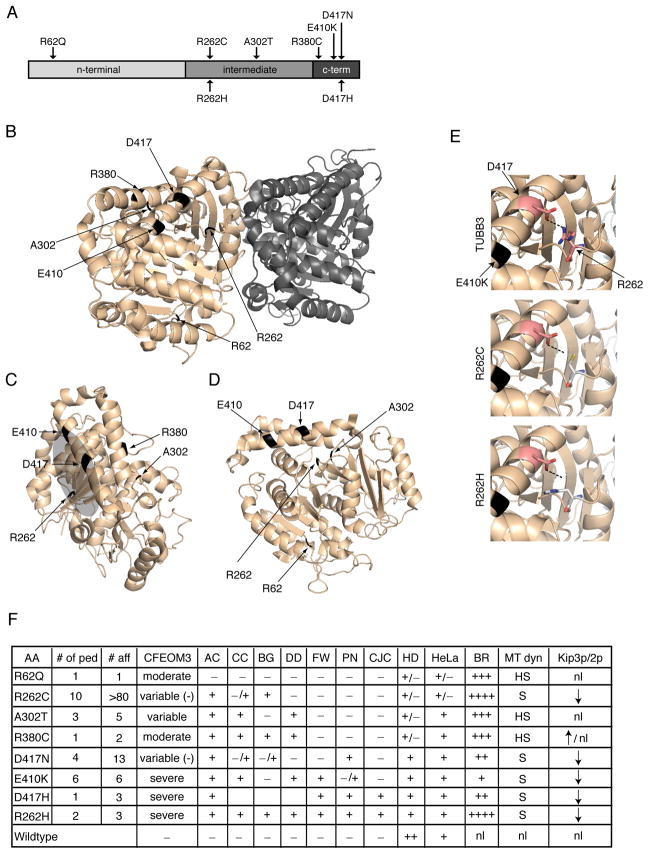
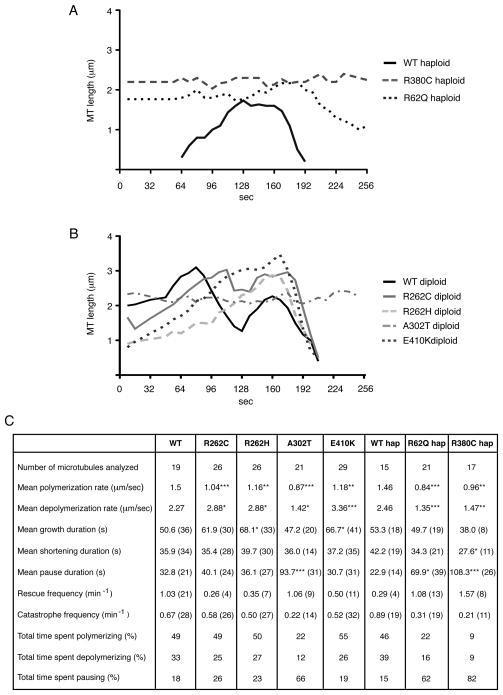
We report that eight heterozygous missense mutations in TUBB3, encoding the neuron-specific beta-tubulin isotype III, result in a spectrum of human nervous system disorders that we now call the TUBB3 syndromes. Each mutation causes the ocular motility disorder CFEOM3, whereas some also result in intellectual and behavioral impairments, facial paralysis, and/or later-onset axonal sensorimotor polyneuropathy. Neuroimaging reveals a spectrum of abnormalities including hypoplasia of oculomotor nerves and dysgenesis of the corpus callosum, anterior commissure, and corticospinal tracts. A knock-in disease mouse model reveals axon guidance defects without evidence of cortical cell migration abnormalities. We show that the disease-associated mutations can impair tubulin heterodimer formation in vitro, although folded mutant heterodimers can still polymerize into microtubules. Modeling each mutation in yeast tubulin demonstrates that all alter dynamic instability whereas a subset disrupts the interaction of microtubules with kinesin motors. These findings demonstrate that normal TUBB3 is required for axon guidance and maintenance in mammals.
我们报道称,TUBB3 基因(编码神经元特异性β-微管蛋白同工型 III)的八个杂合错义突变导致了一系列人类神经系统疾病,我们现在将其称为 TUBB3 综合征。每种突变都会导致眼球运动障碍 CFEOM3,而有些突变还会导致智力和行为障碍、面瘫和/或迟发性轴索性感觉运动多发性神经病。神经影像学显示出一系列异常,包括动眼神经发育不良和胼胝体、前连合和皮质脊髓束发育不良。敲入疾病小鼠模型显示出轴突导向缺陷,而没有皮质细胞迁移异常的证据。我们表明,与疾病相关的突变可以体外损害微管蛋白异二聚体的形成,尽管折叠的突变异二聚体仍能聚合形成微管。在酵母微管蛋白中对每个突变进行建模表明,所有突变都改变了微管的动态不稳定性,而一部分突变则破坏了微管与驱动蛋白的相互作用。这些发现表明,正常的 TUBB3 对于哺乳动物的轴突导向和维持是必需的。