Institut Pasteur, Unité d'Immunologie Moléculaire des Parasites, CNRS URA 2581, 25-28 rue du Dr Roux, 75724, Paris cedex 15, France.
BMC Genomics. 2010 Jan 15;11:34. doi: 10.1186/1471-2164-11-34.
Malaria is the most important parasitic disease in the world with approximately two million people dying every year, mostly due to Plasmodium falciparum infection. During its complex life cycle in the Anopheles vector and human host, the parasite requires the coordinated and modulated expression of diverse sets of genes involved in epigenetic, transcriptional and post-transcriptional regulation. However, despite the availability of the complete sequence of the Plasmodium falciparum genome, we are still quite ignorant about Plasmodium mechanisms of transcriptional gene regulation. This is due to the poor prediction of nuclear proteins, cognate DNA motifs and structures involved in transcription.
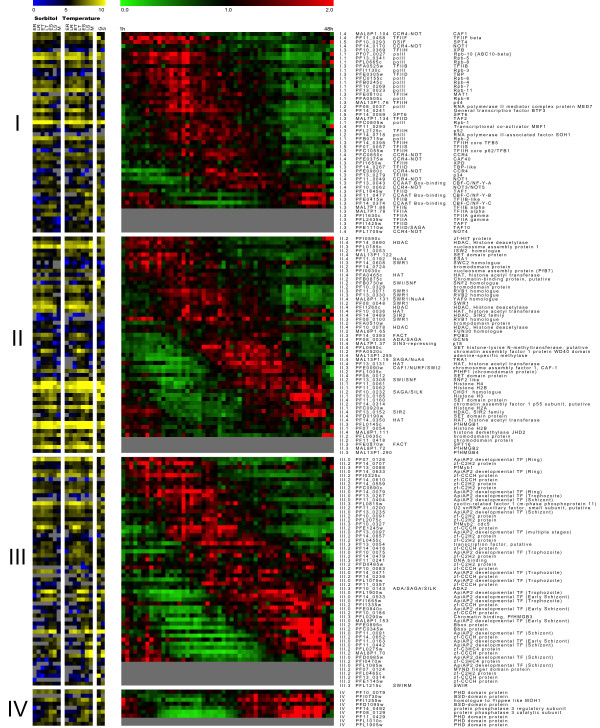
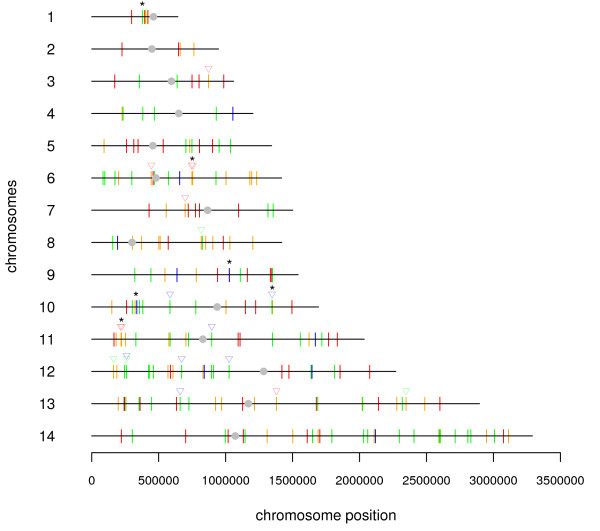
A comprehensive directory of proteins reported to be potentially involved in Plasmodium transcriptional machinery was built from all in silico reports and databanks. The transcription-associated proteins were clustered in three main sets of factors: general transcription factors, chromatin-related proteins (structuring, remodelling and histone modifying enzymes), and specific transcription factors. Only a few of these factors have been molecularly analysed. Furthermore, from transcriptome and proteome data we modelled expression patterns of transcripts and corresponding proteins during the intra-erythrocytic cycle. Finally, an interactome of these proteins based either on in silico or on 2-yeast-hybrid experimental approaches is discussed.
This is the first attempt to build a comprehensive directory of potential transcription-associated proteins in Plasmodium. In addition, all complete transcriptome, proteome and interactome raw data were re-analysed, compared and discussed for a better comprehension of the complex biological processes of Plasmodium falciparum transcriptional regulation during the erythrocytic development.
疟疾是世界上最重要的寄生虫病,每年约有 200 万人死亡,主要是由恶性疟原虫感染引起的。在疟原虫在按蚊媒介和人类宿主中的复杂生命周期中,寄生虫需要协调和调节涉及表观遗传、转录和转录后调节的不同基因集的表达。然而,尽管恶性疟原虫基因组的完整序列已经可用,但我们对疟原虫转录基因调控机制仍然知之甚少。这是由于核蛋白、同源 DNA 基序和转录相关结构的预测能力较差所致。
从所有计算机报告和数据库中构建了一个潜在涉及疟原虫转录机制的蛋白质的综合目录。转录相关蛋白被分为三组主要因子:一般转录因子、染色质相关蛋白(结构、重塑和组蛋白修饰酶)和特异性转录因子。这些因子中只有少数几个进行了分子分析。此外,根据转录组和蛋白质组数据,我们在红细胞内周期期间模拟了转录物和相应蛋白质的表达模式。最后,讨论了基于计算机或双酵母杂交实验方法的这些蛋白质的相互作用组。
这是首次尝试构建恶性疟原虫潜在转录相关蛋白的综合目录。此外,还重新分析、比较和讨论了所有完整的转录组、蛋白质组和相互作用组原始数据,以更好地理解恶性疟原虫红细胞发育过程中转录调控的复杂生物学过程。