Department of Biology, Molecular Neurobiology, University of Oldenburg, Oldenburg, Germany.
PLoS One. 2010 Jan 18;5(1):e8753. doi: 10.1371/journal.pone.0008753.
The accumulation and aggregation of alpha-synuclein in nerve cells and glia are characteristic features of a number of neurodegenerative diseases termed synucleinopathies. alpha-Synuclein is a highly soluble protein which in a nucleation dependent process is capable of self-aggregation. The causes underlying aggregate formation are not yet understood, impairment of the proteolytic degradation systems might be involved.
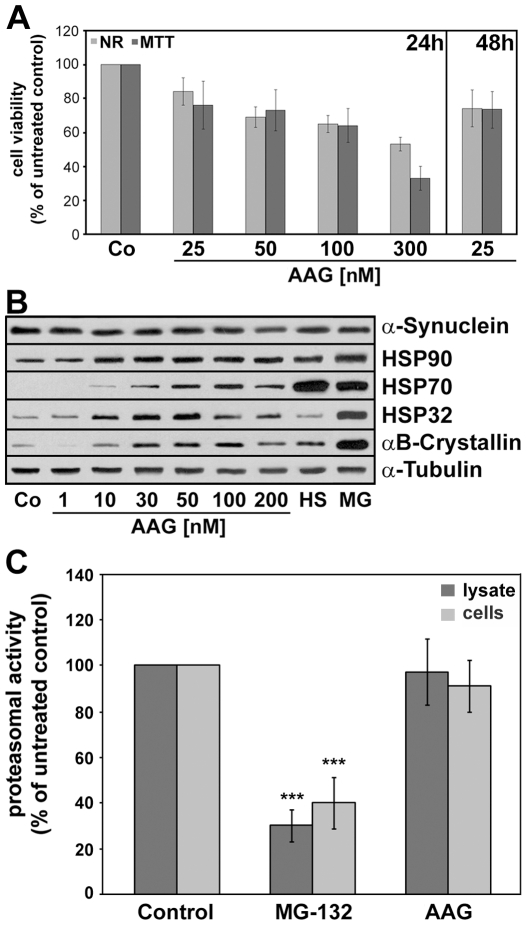
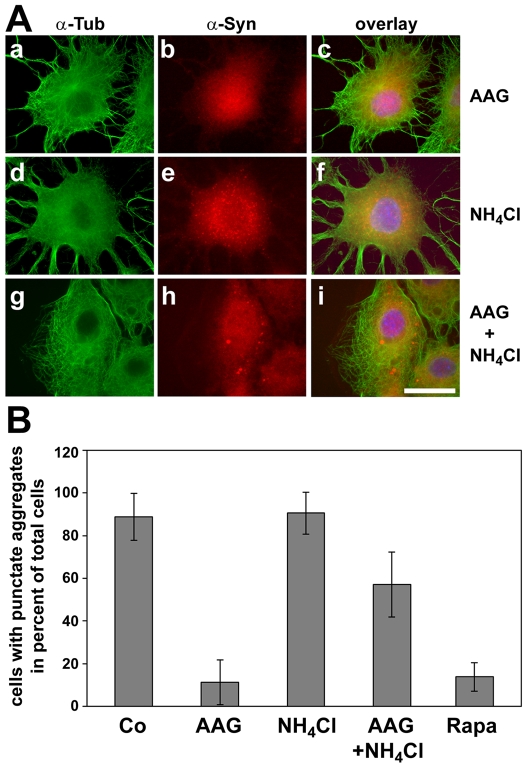
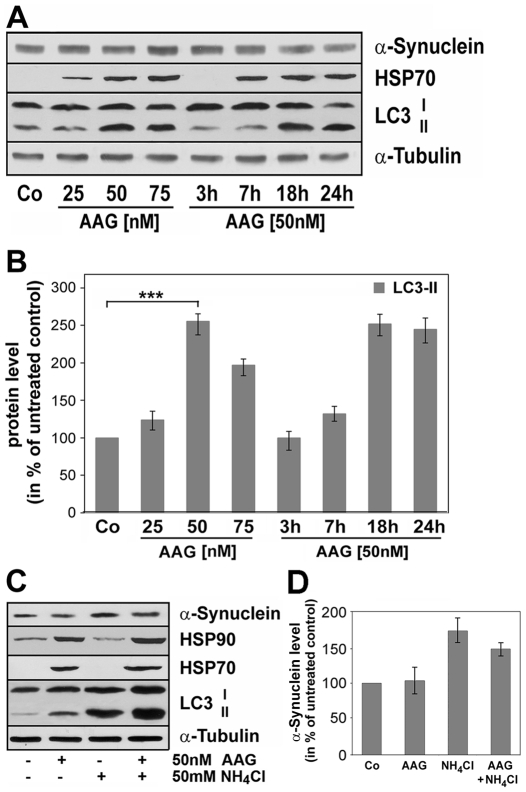
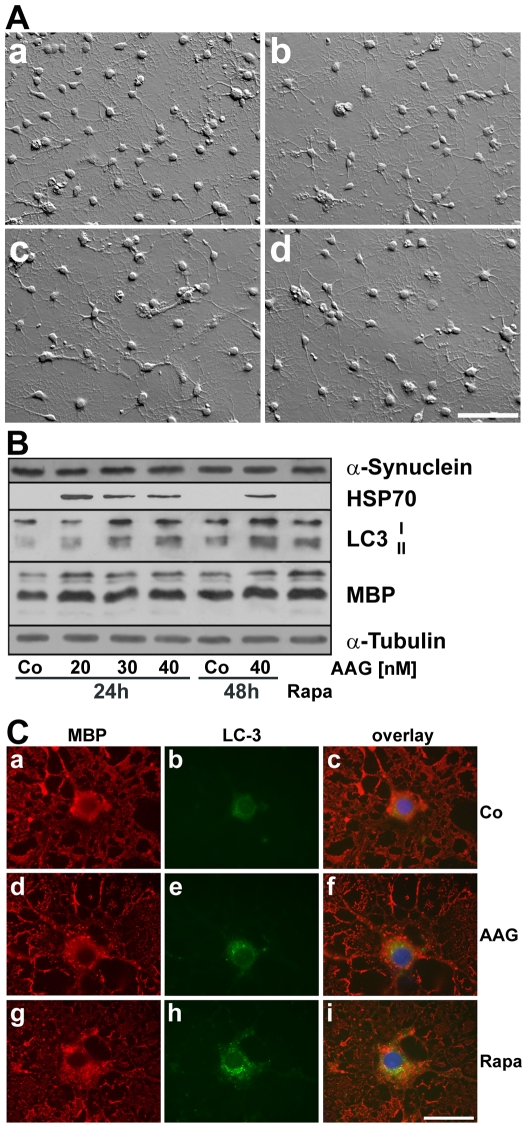
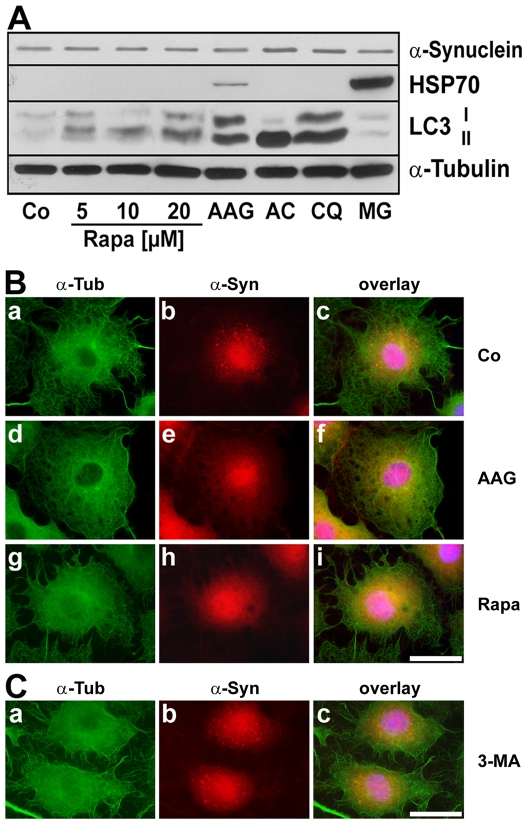
METHODOLOGY/PRINCIPAL FINDINGS: In the present study the possible aggregate clearing effects of the geldanamycin analogue 17-AAG (17-(Allylamino)-17-demethoxygeldanamycin) was investigated. Towards this, an oligodendroglial cell line (OLN-93 cells), stably expressing human alpha-synuclein (A53T mutation) was used. In these cells small punctate aggregates, not staining with thioflavine S, representing prefibrillary aggregates, occur characteristically. Our data demonstrate that 17-AAG attenuated the formation of alpha-synuclein aggregates by stimulating macroautophagy. By blocking the lysosomal compartment with NH(4)Cl the aggregate clearing effects of 17-AAG were abolished and alpha-synuclein deposits were enlarged. Analysis of LC3-II immunoreactivity, which is an indicator of autophagosome formation, further revealed that 17-AAG led to the recruitment of LC3-II and to the formation of LC3 positive puncta. This effect was also observed in cultured oligodendrocytes derived from the brains of newborn rats. Inhibition of macroautophagy by 3-methyladenine prevented 17-AAG induced occurrence of LC3 positive puncta as well as the removal of alpha-synuclein aggregates in OLN-A53T cells.
Our data demonstrate for the first time that 17-AAG not only causes the upregulation of heat shock proteins, but also is an effective inducer of the autophagic pathway by which alpha-synuclein can be removed. Hence geldanamycin derivatives may provide a means to modulate autophagy in neural cells, thereby ameliorating pathogenic aggregate formation and protecting the cells during disease and aging.
α-突触核蛋白在神经细胞和神经胶质细胞中的积累和聚集是几种神经退行性疾病(称为突触核蛋白病)的特征。α-突触核蛋白是一种高度可溶性的蛋白质,在依赖成核的过程中能够自我聚集。聚合形成的原因尚不清楚,可能涉及到蛋白酶体降解系统的损伤。
方法/主要发现:在本研究中,研究了格尔德霉素类似物 17-AAG(17-(烯丙基氨基)-17-去甲氧基格尔德霉素)的可能的聚集清除作用。为此,使用了稳定表达人α-突触核蛋白(A53T 突变)的少突胶质细胞系(OLN-93 细胞)。在这些细胞中,会出现特征性的小而点状的聚集物,这些聚集物不与硫黄素 S 染色,代表前纤维状聚集物。我们的数据表明,17-AAG 通过刺激巨自噬来减弱α-突触核蛋白聚集的形成。用氯化铵阻断溶酶体区室,17-AAG 的聚集清除作用被消除,α-突触核蛋白沉积物增大。分析 LC3-II 免疫反应性,这是自噬体形成的一个指标,进一步表明 17-AAG 导致 LC3-II 的募集和 LC3 阳性点状结构的形成。这一效应也在新生大鼠脑源性培养少突胶质细胞中观察到。用 3-甲基腺嘌呤抑制巨自噬,可防止 17-AAG 诱导的 LC3 阳性点状结构的形成以及 OLN-A53T 细胞中α-突触核蛋白聚集物的清除。
我们的数据首次表明,17-AAG 不仅能引起热休克蛋白的上调,而且还能有效地诱导自噬途径,从而清除α-突触核蛋白。因此,格尔德霉素衍生物可能为调节神经细胞中的自噬提供一种手段,从而改善致病聚集物的形成,并在疾病和衰老过程中保护细胞。