MCRI Center for Translational Genomics, Molecular Cardiology Research Institute, Boston, Massachusetts, United States of America.
PLoS One. 2010 Jan 21;5(1):e8830. doi: 10.1371/journal.pone.0008830.
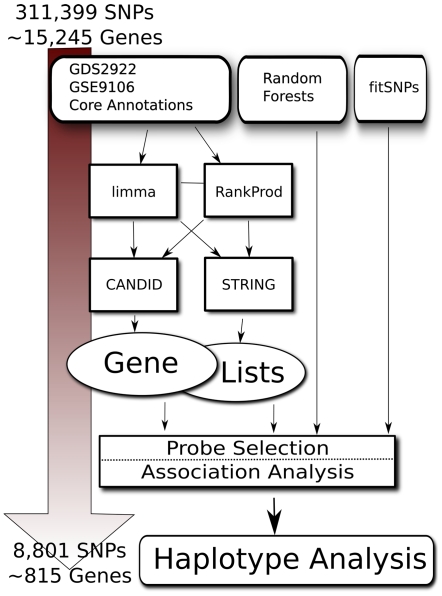
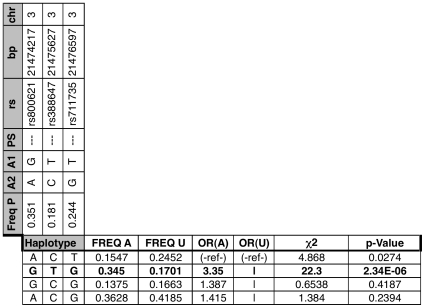
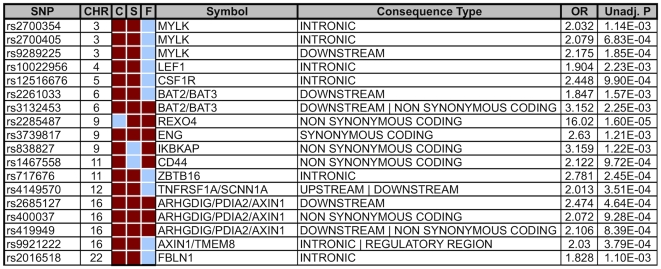
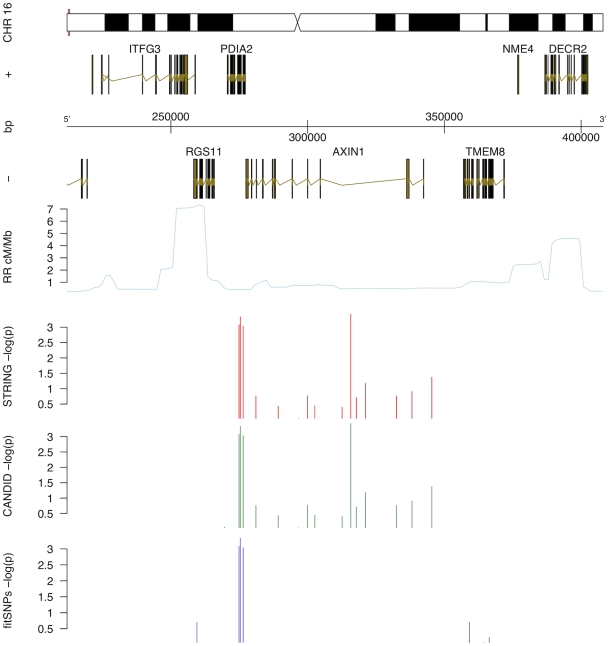
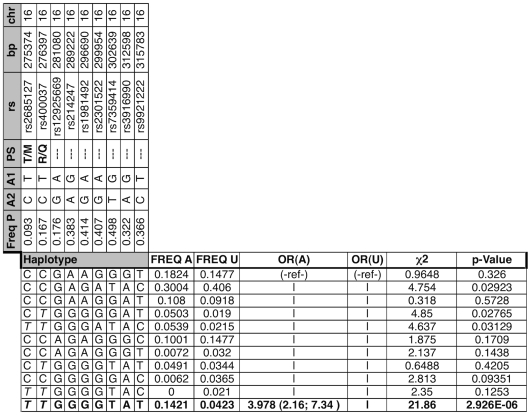
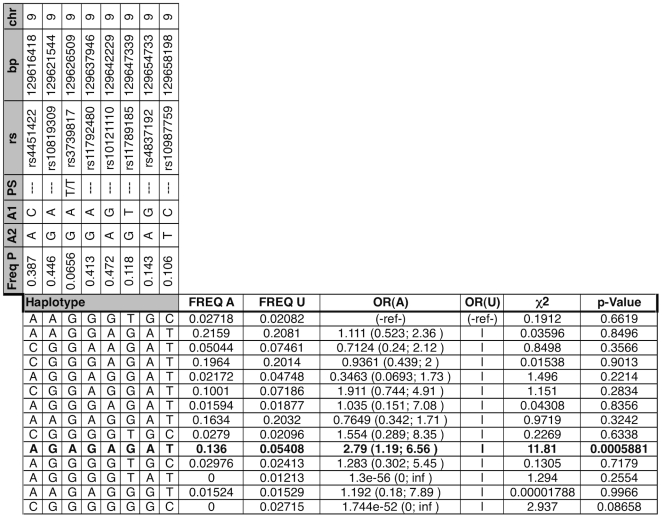
Bicuspid Aortic Valve (BAV) is a highly heritable congenital heart defect. The low frequency of BAV (1% of general population) limits our ability to perform genome-wide association studies. We present the application of four a priori SNP selection techniques, reducing the multiple-testing penalty by restricting analysis to SNPs relevant to BAV in a genome-wide SNP dataset from a cohort of 68 BAV probands and 830 control subjects. Two knowledge-based approaches, CANDID and STRING, were used to systematically identify BAV genes, and their SNPs, from the published literature, microarray expression studies and a genome scan. We additionally tested Functionally Interpolating SNPs (fitSNPs) present on the array; the fourth consisted of SNPs selected by Random Forests, a machine learning approach. These approaches reduced the multiple testing penalty by lowering the fraction of the genome probed to 0.19% of the total, while increasing the likelihood of studying SNPs within relevant BAV genes and pathways. Three loci were identified by CANDID, STRING, and fitSNPS. A haplotype within the AXIN1-PDIA2 locus (p-value of 2.926x10(-06)) and a haplotype within the Endoglin gene (p-value of 5.881x10(-04)) were found to be strongly associated with BAV. The Random Forests approach identified a SNP on chromosome 3 in association with BAV (p-value 5.061x10(-06)). The results presented here support an important role for genetic variants in BAV and provide support for additional studies in well-powered cohorts. Further, these studies demonstrate that leveraging existing expression and genomic data in the context of GWAS studies can identify biologically relevant genes and pathways associated with a congenital heart defect.
二叶式主动脉瓣(BAV)是一种高度遗传性先天性心脏缺陷。BAV 的低频率(占总人口的 1%)限制了我们进行全基因组关联研究的能力。我们提出了四种先验 SNP 选择技术的应用,通过将分析限制在与 68 名 BAV 先证者和 830 名对照者的全基因组 SNP 数据集相关的 SNP 上,减少了多重检验的惩罚。两种基于知识的方法,CANDID 和 STRING,被用于从已发表的文献、微阵列表达研究和基因组扫描中系统地识别 BAV 基因及其 SNP。我们还测试了功能插值 SNP(fitSNP)在阵列上的存在;第四种方法由随机森林(一种机器学习方法)选择的 SNP 组成。这些方法通过将探测基因组的比例降低到总基因组的 0.19%,同时增加了研究与 BAV 相关基因和途径内 SNP 的可能性,从而降低了多重检验的惩罚。通过 CANDID、STRING 和 fitSNP 方法鉴定了三个基因座。AXIN1-PDIA2 基因座内的单倍型(p 值为 2.926x10(-06)) 和内胚层基因内的单倍型(p 值为 5.881x10(-04)) 与 BAV 强烈相关。随机森林方法确定了与 BAV 相关的 3 号染色体上的 SNP(p 值为 5.061x10(-06))。这里提出的结果支持遗传变异在 BAV 中的重要作用,并为在有足够能力的队列中进行更多研究提供了支持。此外,这些研究表明,在 GWAS 研究的背景下利用现有的表达和基因组数据可以识别与先天性心脏缺陷相关的生物学上相关的基因和途径。