J, Craig Venter Institute, 9704 Medical Center Drive, Rockville, MD 20850, USA.
BMC Bioinformatics. 2010 Jan 26;11:52. doi: 10.1186/1471-2105-11-52.
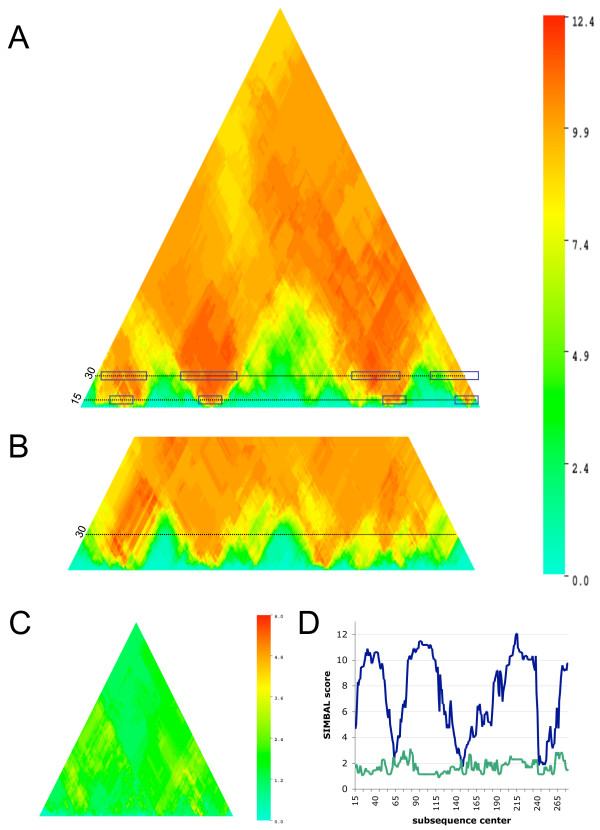
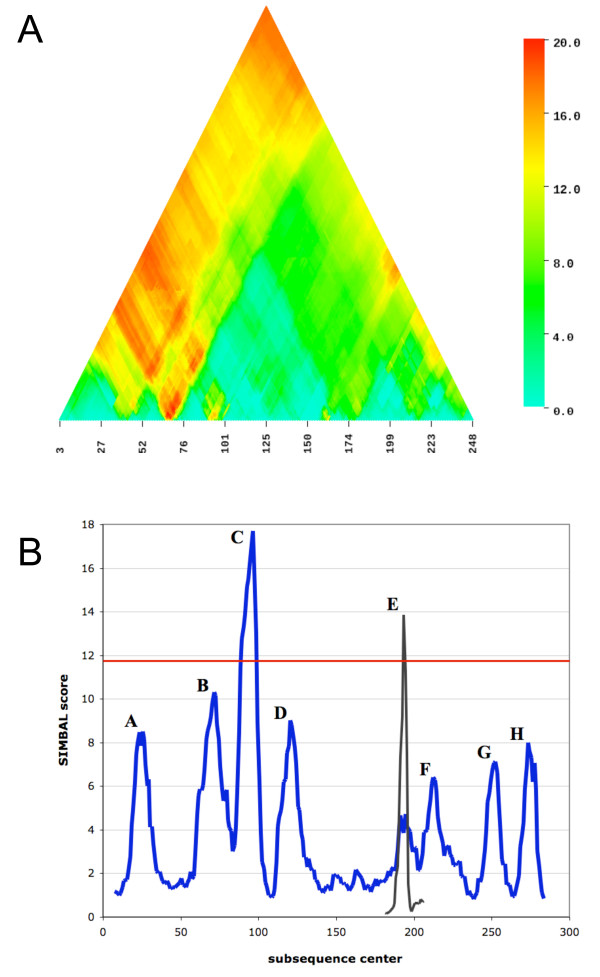
Comparative genomics methods such as phylogenetic profiling can mine powerful inferences from inherently noisy biological data sets. We introduce Sites Inferred by Metabolic Background Assertion Labeling (SIMBAL), a method that applies the Partial Phylogenetic Profiling (PPP) approach locally within a protein sequence to discover short sequence signatures associated with functional sites. The approach is based on the basic scoring mechanism employed by PPP, namely the use of binomial distribution statistics to optimize sequence similarity cutoffs during searches of partitioned training sets.
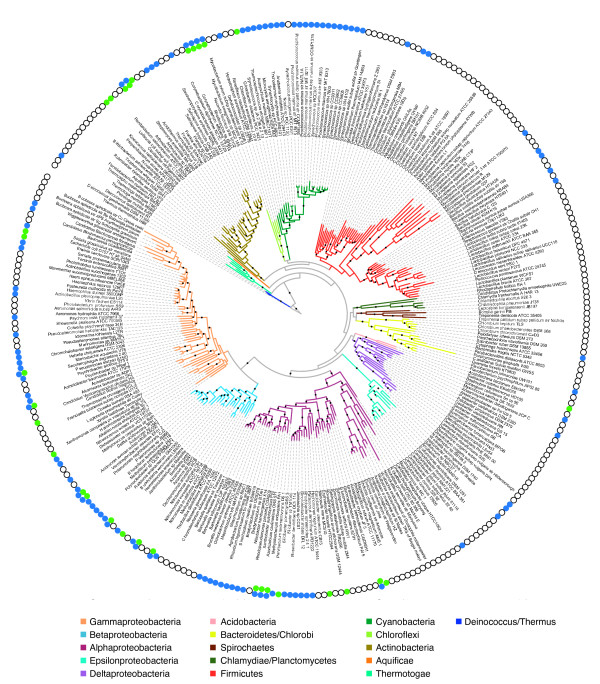
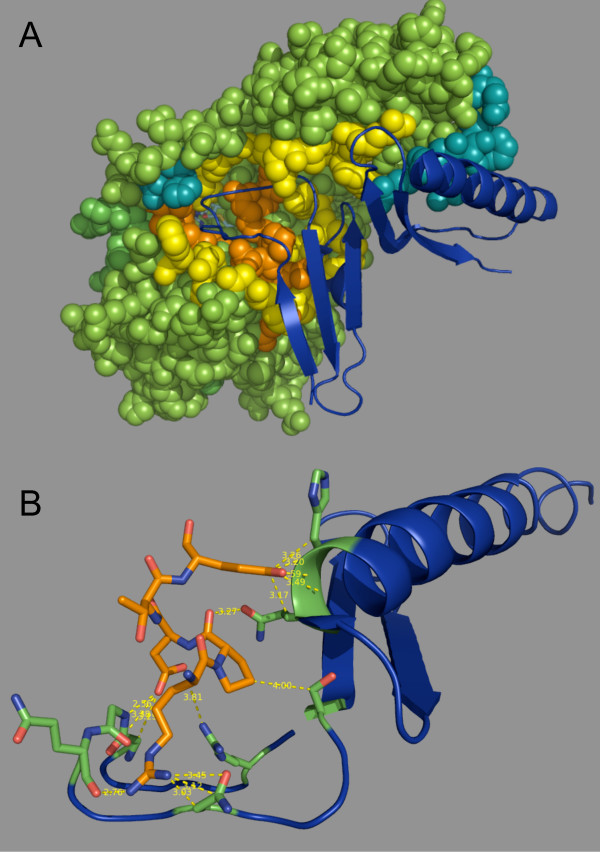
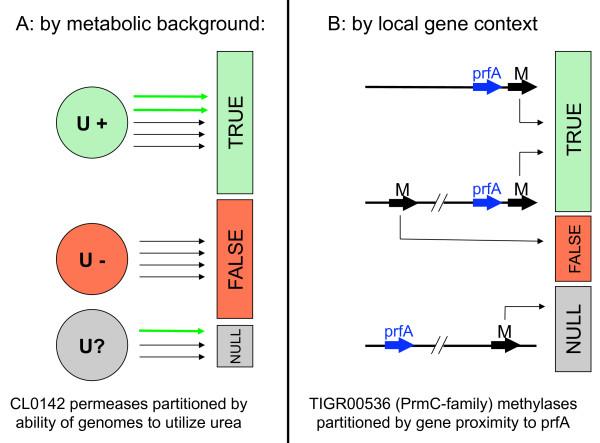
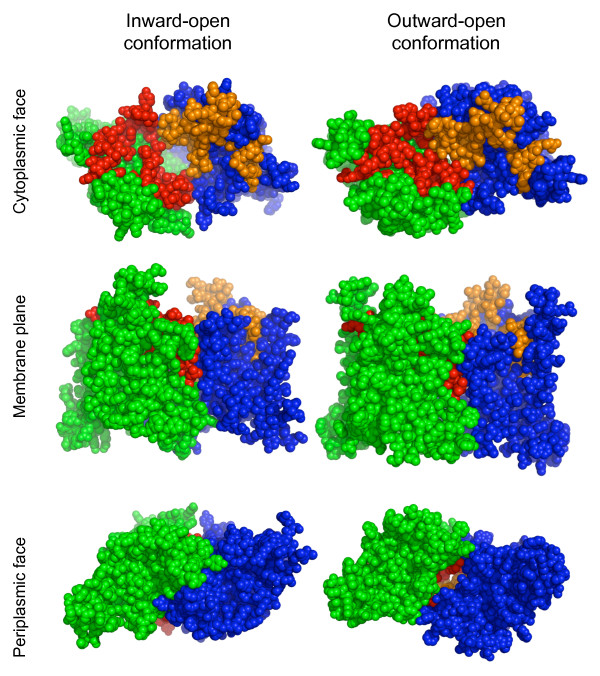
Here we illustrate and validate the ability of the SIMBAL method to find functionally relevant short sequence signatures by application to two well-characterized protein families. In the first example, we partitioned a family of ABC permeases using a metabolic background property (urea utilization). Thus, the TRUE set for this family comprised members whose genome of origin encoded a urea utilization system. By moving a sliding window across the sequence of a permease, and searching each subsequence in turn against the full set of partitioned proteins, the method found which local sequence signatures best correlated with the urea utilization trait. Mapping of SIMBAL "hot spots" onto crystal structures of homologous permeases reveals that the significant sites are gating determinants on the cytosolic face rather than, say, docking sites for the substrate-binding protein on the extracellular face. In the second example, we partitioned a protein methyltransferase family using gene proximity as a criterion. In this case, the TRUE set comprised those methyltransferases encoded near the gene for the substrate RF-1. SIMBAL identifies sequence regions that map onto the substrate-binding interface while ignoring regions involved in the methyltransferase reaction mechanism in general. Neither method for training set construction requires any prior experimental characterization.
SIMBAL shows that, in functionally divergent protein families, selected short sequences often significantly outperform their full-length parent sequence for making functional predictions by sequence similarity, suggesting avenues for improved functional classifiers. When combined with structural data, SIMBAL affords the ability to localize and model functional sites.
比较基因组学方法,如系统发育分析,可以从固有噪声的生物数据集挖掘出强大的推论。我们引入了代谢背景断言标记推断的位点(SIMBAL),这是一种在蛋白质序列内局部应用部分系统发育分析(PPP)方法来发现与功能位点相关的短序列特征的方法。该方法基于 PPP 采用的基本评分机制,即使用二项式分布统计来优化搜索分区训练集时的序列相似性截止值。
在这里,我们通过应用于两个具有良好特征的蛋白质家族来说明和验证 SIMBAL 方法发现功能相关短序列特征的能力。在第一个例子中,我们使用代谢背景特性(尿素利用)来划分 ABC 转运体家族。因此,该家族的 TRUE 集由其起源基因组编码尿素利用系统的成员组成。通过在转运体的序列上移动滑动窗口,并依次搜索每个子序列与完整的分区蛋白集,该方法找到了与尿素利用特性最相关的局部序列特征。将 SIMBAL“热点”映射到同源转运体的晶体结构上表明,显著的位点是细胞质侧的门控决定因素,而不是细胞外侧的底物结合蛋白的停靠位点。在第二个例子中,我们使用基因邻近性作为标准来划分蛋白质甲基转移酶家族。在这种情况下,TRUE 集由那些编码在底物 RF-1 基因附近的甲基转移酶组成。SIMBAL 识别映射到底物结合界面的序列区域,同时忽略一般涉及甲基转移酶反应机制的区域。两种训练集构建方法都不需要任何先前的实验表征。
SIMBAL 表明,在功能上不同的蛋白质家族中,选择的短序列通常比其全长序列更能通过序列相似性进行功能预测,这为改进功能分类器提供了途径。当与结构数据结合使用时,SIMBAL 可以定位和模拟功能位点。