Cheng Haoyu, Jiang Lihua, Wu Maoying, Liu Qi
School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai 200240, China. Email:
Bioinform Biol Insights. 2009 Oct 21;3:129-40. doi: 10.4137/bbi.s3445.
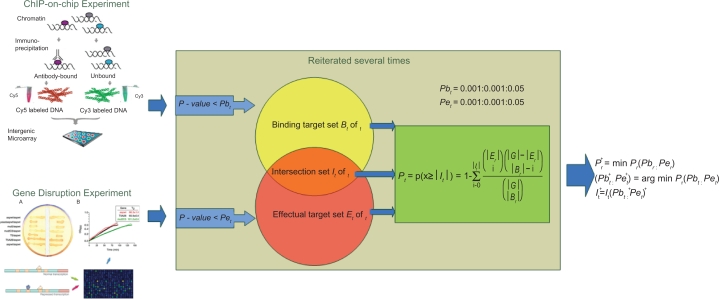
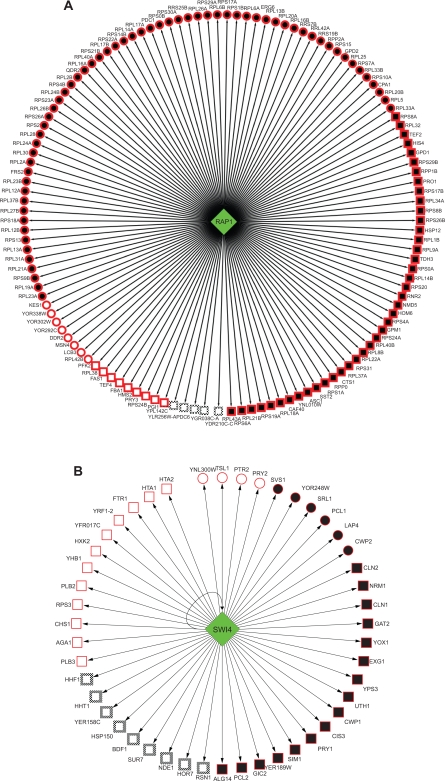
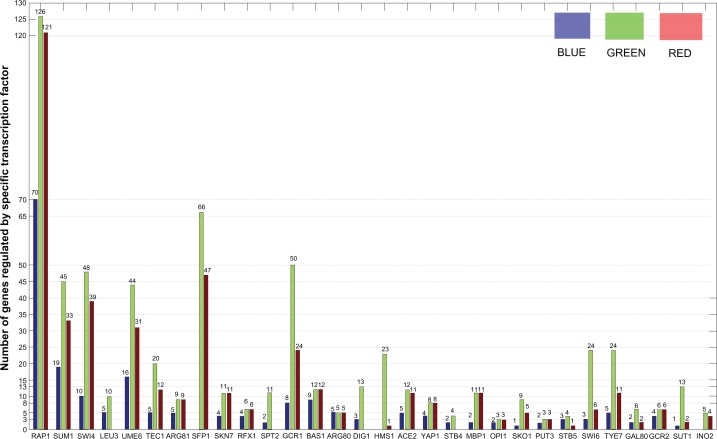
How to combine heterogeneous data sources for reliable prediction of transcriptional regulation is a challenge. Here we present an easy but powerful method to integrate Chromatin immunoprecipitation (ChIP)-chip and knock-out data. Since these two types of data provide complementary (physical and functional) information about transcription, the method combining them is expected to achieve high detection rates and very low false positive rates. We try to seek the optimal integration of these two data using hyper-geometric distribution. We evaluate our method on yeast data and compare our predictions with YEASTRACT, high-quality ChIP-chip data, and literature. The results show that even using low-quality ChIP-chip data, our method uncovers more relations than those inferred before from high-quality data. Furthermore our method achieves a low false positive rate. We find experimental and computational evidence in literature for most transcription factor (TF)-gene relations uncovered by our method.
如何整合异构数据源以可靠地预测转录调控是一项挑战。在此,我们提出一种简单但强大的方法来整合染色质免疫沉淀(ChIP)芯片和基因敲除数据。由于这两种类型的数据提供了关于转录的互补(物理和功能)信息,因此将它们结合起来的方法有望实现高检测率和极低的假阳性率。我们尝试使用超几何分布来寻求这两种数据的最佳整合。我们在酵母数据上评估了我们的方法,并将我们的预测与YEASTRACT、高质量的ChIP芯片数据和文献进行了比较。结果表明,即使使用低质量的ChIP芯片数据,我们的方法也能发现比以前从高质量数据中推断出的更多关系。此外,我们的方法实现了低假阳性率。我们在文献中找到了实验和计算证据,证明了我们的方法所发现的大多数转录因子(TF)-基因关系。