Center for Human Genetics, Duke University Medical Center, Durham, NC 27710, USA.
Nucleic Acids Res. 2010 May;38(9):e105. doi: 10.1093/nar/gkq040. Epub 2010 Feb 8.
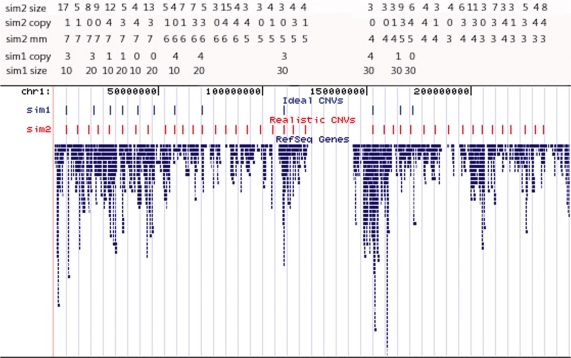
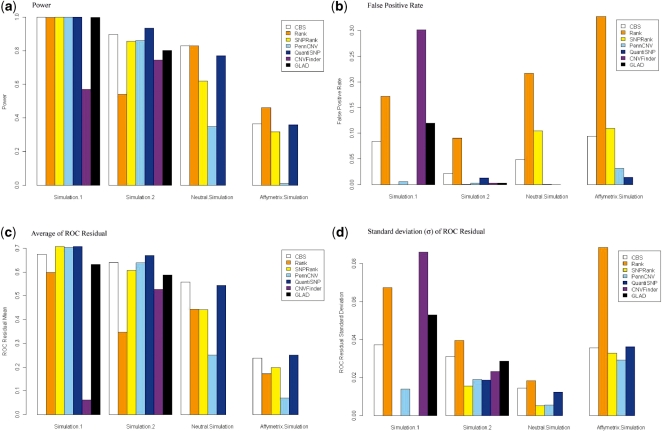
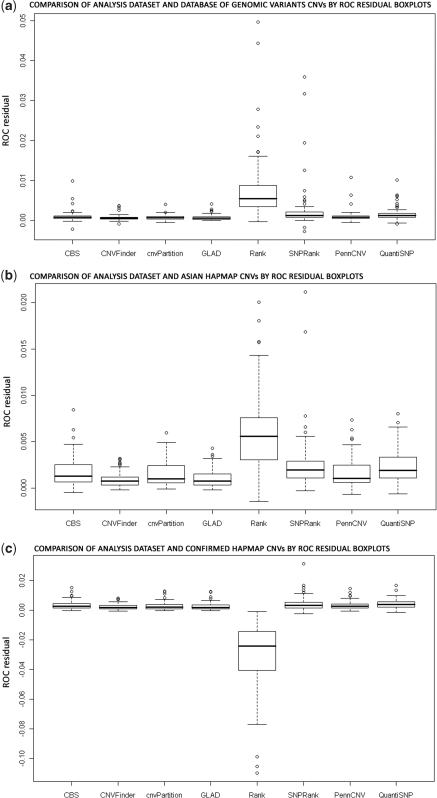
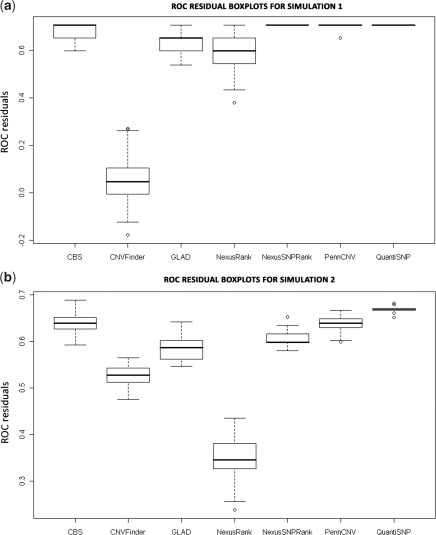
Determination of copy number variants (CNVs) inferred in genome wide single nucleotide polymorphism arrays has shown increasing utility in genetic variant disease associations. Several CNV detection methods are available, but differences in CNV call thresholds and characteristics exist. We evaluated the relative performance of seven methods: circular binary segmentation, CNVFinder, cnvPartition, gain and loss of DNA, Nexus algorithms, PennCNV and QuantiSNP. Tested data included real and simulated Illumina HumHap 550 data from the Singapore cohort study of the risk factors for Myopia (SCORM) and simulated data from Affymetrix 6.0 and platform-independent distributions. The normalized singleton ratio (NSR) is proposed as a metric for parameter optimization before enacting full analysis. We used 10 SCORM samples for optimizing parameter settings for each method and then evaluated method performance at optimal parameters using 100 SCORM samples. The statistical power, false positive rates, and receiver operating characteristic (ROC) curve residuals were evaluated by simulation studies. Optimal parameters, as determined by NSR and ROC curve residuals, were consistent across datasets. QuantiSNP outperformed other methods based on ROC curve residuals over most datasets. Nexus Rank and SNPRank have low specificity and high power. Nexus Rank calls oversized CNVs. PennCNV detects one of the fewest numbers of CNVs.
全基因组单核苷酸多态性微阵列推断的拷贝数变异 (CNV) 在遗传变异疾病关联中的应用越来越广泛。有几种 CNV 检测方法,但 CNV 调用阈值和特征存在差异。我们评估了七种方法的相对性能:圆形二进制分割、CNVFinder、cnvPartition、DNA 增益和丢失、Nexus 算法、PennCNV 和 QuantiSNP。测试数据包括来自新加坡近视危险因素研究 (SCORM) 的真实和模拟 Illumina HumHap 550 数据以及来自 Affymetrix 6.0 和与平台无关分布的模拟数据。归一化单核苷酸比 (NSR) 被提议作为在执行全面分析之前优化参数的指标。我们使用 10 个 SCORM 样本对每种方法的参数进行了优化设置,然后使用 100 个 SCORM 样本在最佳参数下评估了方法性能。通过模拟研究评估了统计功效、假阳性率和接收器工作特征 (ROC) 曲线残差。通过 NSR 和 ROC 曲线残差确定的最佳参数在不同数据集之间是一致的。基于 ROC 曲线残差,QuantiSNP 在大多数数据集上的性能优于其他方法。Nexus Rank 和 SNPRank 的特异性低,功效高。Nexus Rank 调用过大的 CNV。PennCNV 检测到的 CNV 数量最少。