Department of Oral Biology, Faculty of Dentistry, University of Oslo, Oslo, Norway.
BMC Bioinformatics. 2010 Feb 9;11:82. doi: 10.1186/1471-2105-11-82.
Current commercial high-density oligonucleotide microarrays can hold millions of probe spots on a single microscopic glass slide and are ideal for studying the transcriptome of microbial genomes using a tiling probe design. This paper describes a comprehensive computational pipeline implemented specifically for designing tiling probe sets to study microbial transcriptome profiles.
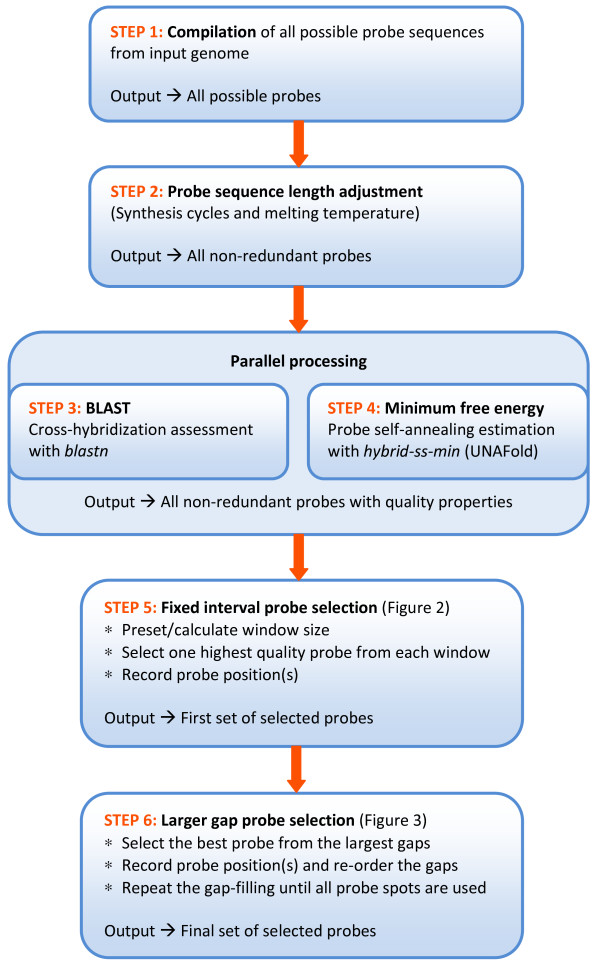
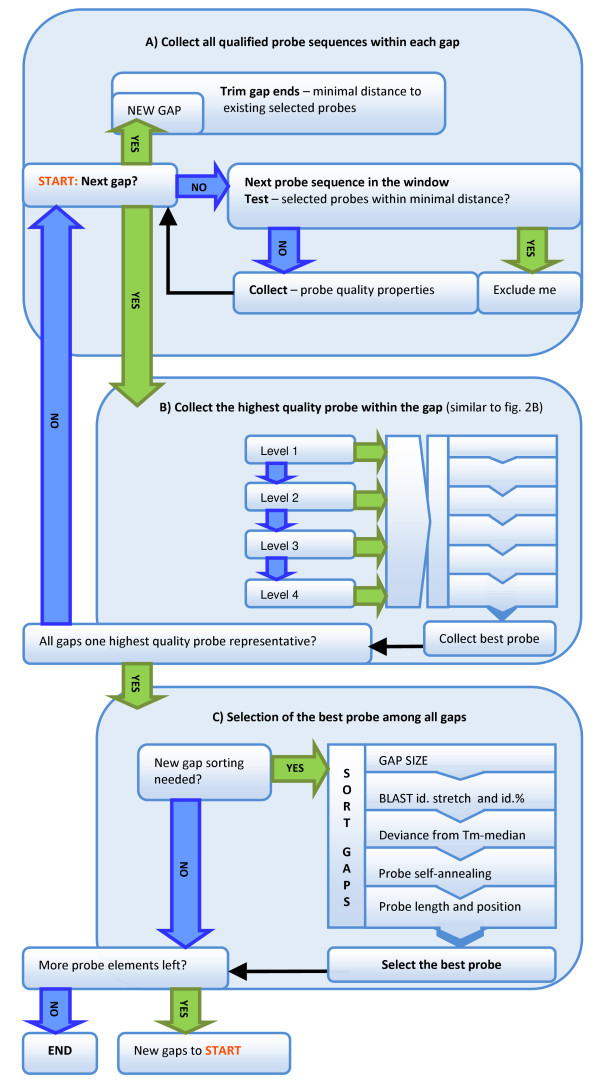
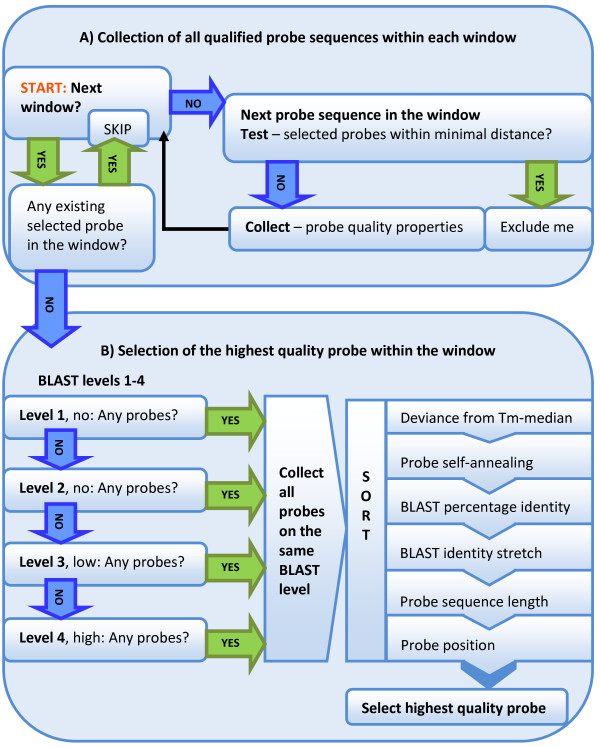
The pipeline identifies every possible probe sequence from both forward and reverse-complement strands of all DNA sequences in the target genome including circular or linear chromosomes and plasmids. Final probe sequence lengths are adjusted based on the maximal oligonucleotide synthesis cycles and best isothermality allowed. Optimal probes are then selected in two stages - sequential and gap-filling. In the sequential stage, probes are selected from sequence windows tiled alongside the genome. In the gap-filling stage, additional probes are selected from the largest gaps between adjacent probes that have already been selected, until a predefined number of probes is reached. Selection of the highest quality probe within each window and gap is based on five criteria: sequence uniqueness, probe self-annealing, melting temperature, oligonucleotide length, and probe position.
The probe selection pipeline evaluates global and local probe sequence properties and selects a set of probes dynamically and evenly distributed along the target genome. Unique to other similar methods, an exact number of non-redundant probes can be designed to utilize all the available probe spots on any chosen microarray platform. The pipeline can be applied to microbial genomes when designing high-density tiling arrays for comparative genomics, ChIP chip, gene expression and comprehensive transcriptome studies.
目前的商业高密度寡核苷酸微阵列可以在单个显微镜载玻片上容纳数百万个探针点,非常适合使用平铺探针设计研究微生物基因组的转录组。本文描述了一个专门用于设计平铺探针组以研究微生物转录组图谱的综合计算流程。
该流程从目标基因组中所有 DNA 序列的正向和反向互补链中识别出每一个可能的探针序列,包括圆形或线性染色体和质粒。最终探针序列长度根据最大寡核苷酸合成循环和允许的最佳等温性进行调整。然后在两个阶段进行最佳探针的选择 - 顺序和间隙填充。在顺序阶段,从沿基因组平铺的序列窗口中选择探针。在间隙填充阶段,从已经选择的相邻探针之间的最大间隙中选择额外的探针,直到达到预定的探针数量。在每个窗口和间隙内选择最高质量的探针是基于五个标准:序列独特性、探针自我退火、熔点、寡核苷酸长度和探针位置。
探针选择流程评估全局和局部探针序列特性,并动态选择一组均匀分布在目标基因组上的探针。与其他类似方法不同的是,可以设计出确切数量的非冗余探针,以利用任何选定的微阵列平台上的所有可用探针点。当为比较基因组学、ChIP 芯片、基因表达和综合转录组研究设计高密度平铺阵列时,可以将该流程应用于微生物基因组。